Division of Nephrology, Department of Medicine, Duke University Health System, Durham, North Carolina, USA.
Division of Nephrology, Department of Medicine, Columbia University College of Physicians and Surgeons, New York, New York, USA.
Kidney Int. 2019 Feb;95(2):321-332. doi: 10.1016/j.kint.2018.09.026.
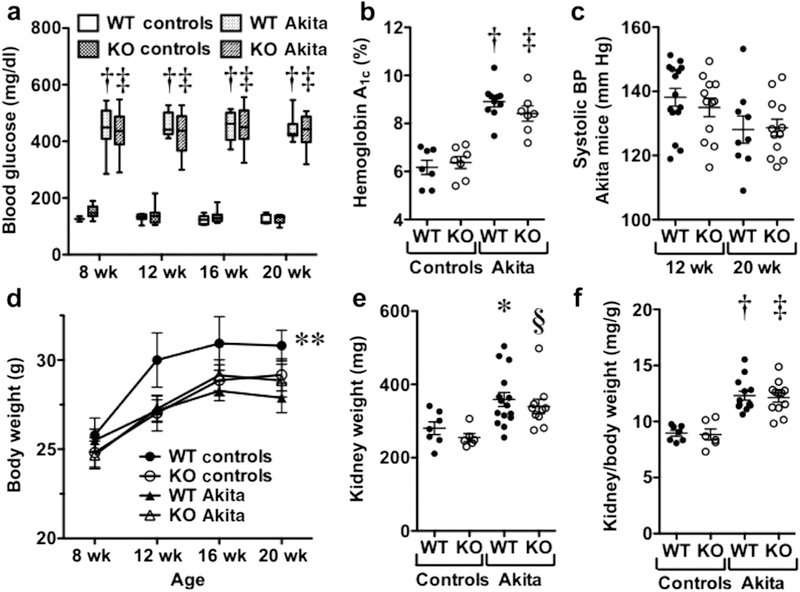
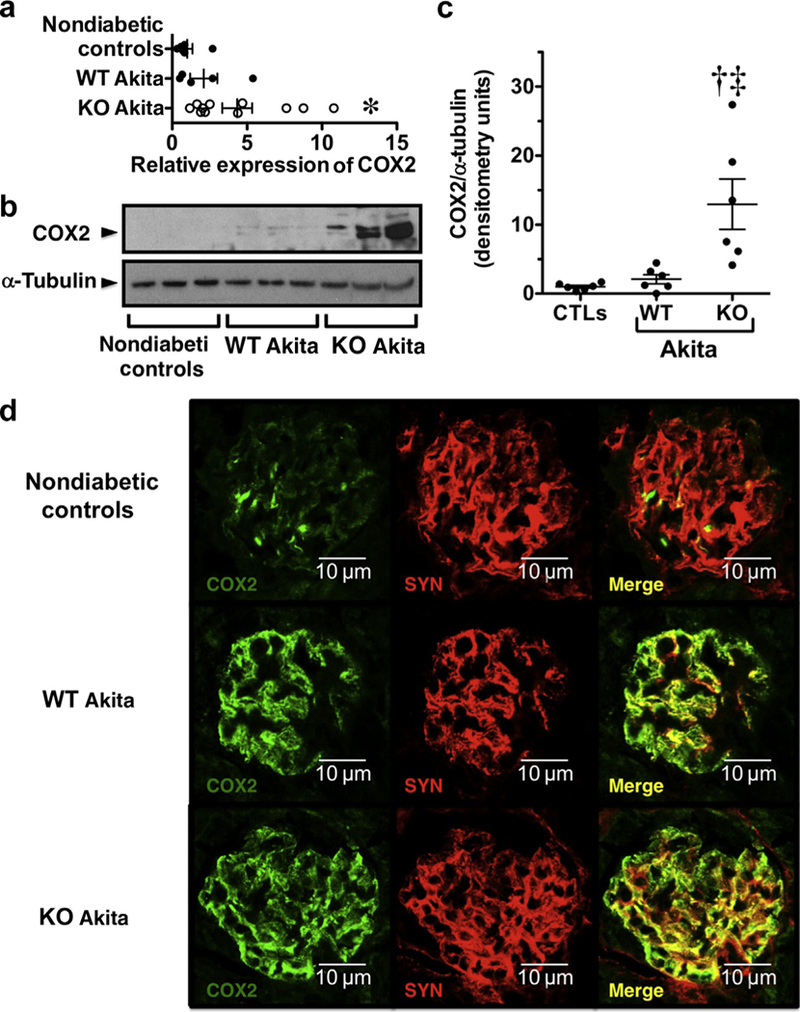
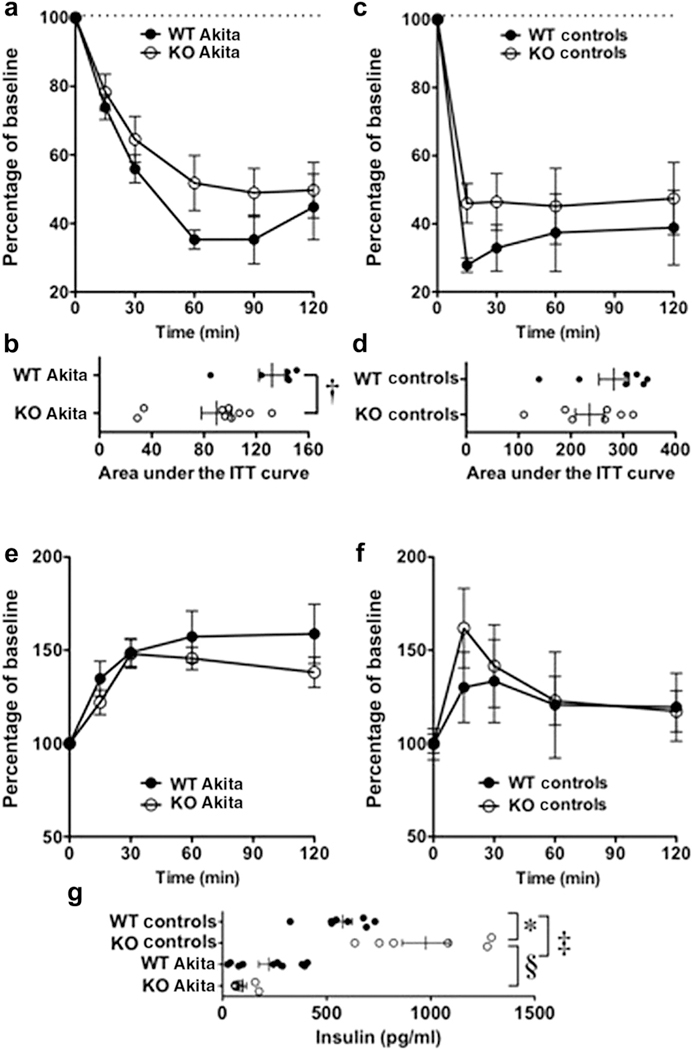
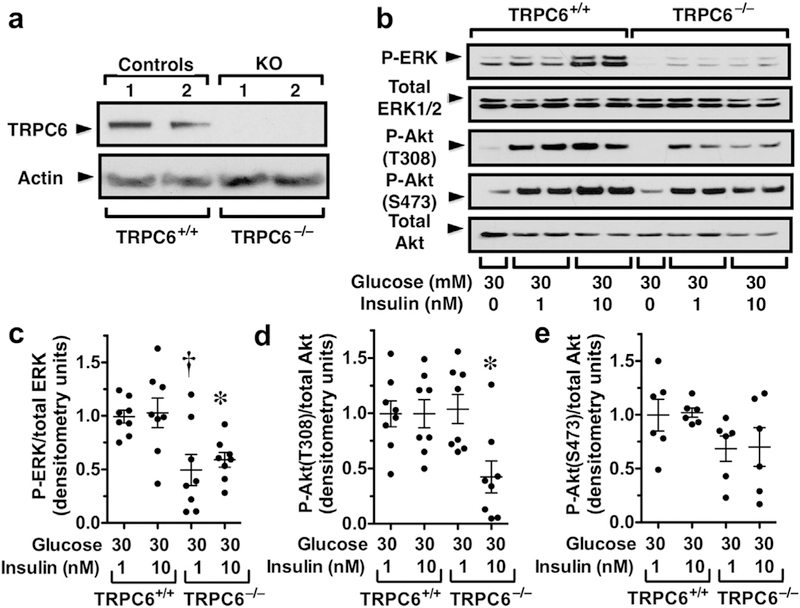
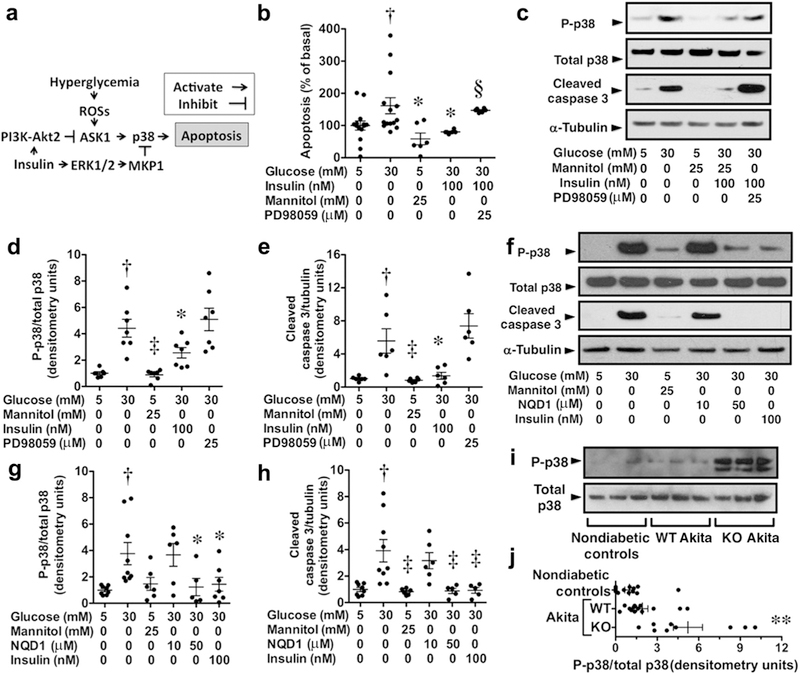
Gain-of-function mutations in TRPC6 cause familial focal segmental glomerulosclerosis, and TRPC6 is upregulated in glomerular diseases including diabetic kidney disease. We studied the effect of systemic TRPC6 knockout in the Akita model of type 1 diabetes. Knockout of TRPC6 inhibited albuminuria in Akita mice at 12 and 16 weeks of age, but this difference disappeared by 20 weeks. Knockout of TRPC6 also reduced tubular injury in Akita mice; however, mesangial expansion was significantly increased. Hyperglycemia and blood pressure were similar between TRPC6 knockout and wild-type Akita mice, but knockout mice were more insulin resistant. In cultured podocytes, knockout of TRPC6 inhibited expression of the calcium/calcineurin responsive gene insulin receptor substrate 2 and decreased insulin responsiveness. Insulin resistance is reported to promote diabetic kidney disease independent of blood glucose levels. While the mechanisms are not fully understood, insulin activates both Akt2 and ERK, which inhibits apoptosis signal regulated kinase 1 (ASK1)-p38-induced apoptosis. In cultured podocytes, hyperglycemia stimulated p38 signaling and induced apoptosis, which was reduced by insulin and ASK1 inhibition and enhanced by Akt or ERK inhibition. Glomerular p38 signaling was increased in TRPC6 knockout Akita mice and was associated with enhanced expression of the p38 gene target cyclooxygenase 2. These data suggest that knockout of TRPC6 in Akita mice promotes insulin resistance and exacerbates glomerular disease independent of hyperglycemia.
TRPC6 的功能获得性突变可导致家族性局灶节段性肾小球硬化症,而 TRPC6 在包括糖尿病肾病在内的多种肾小球疾病中表达上调。我们研究了在 1 型糖尿病 Akita 模型中系统性敲除 TRPC6 的效果。在 12 周和 16 周龄的 Akita 小鼠中,敲除 TRPC6 可抑制白蛋白尿,但到 20 周时这种差异消失。敲除 TRPC6 还可减少 Akita 小鼠的肾小管损伤;然而,系膜扩张显著增加。TRPC6 敲除和野生型 Akita 小鼠的血糖和血压相似,但敲除小鼠的胰岛素抵抗更严重。在培养的足细胞中,敲除 TRPC6 可抑制钙/钙调神经磷酸酶反应基因胰岛素受体底物 2 的表达,并降低胰岛素反应性。据报道,胰岛素抵抗可促进糖尿病肾病的发生,而与血糖水平无关。虽然其机制尚未完全阐明,但胰岛素激活 Akt2 和 ERK,后者可抑制凋亡信号调节激酶 1(ASK1)-p38 诱导的细胞凋亡。在培养的足细胞中,高血糖可刺激 p38 信号通路并诱导细胞凋亡,而胰岛素和 ASK1 抑制可减少这种凋亡,Akt 或 ERK 抑制则可增强这种凋亡。TRPC6 敲除 Akita 小鼠的肾小球 p38 信号增加,与 p38 基因靶标环氧化酶 2 的表达增强有关。这些数据表明,在 Akita 小鼠中敲除 TRPC6 可促进胰岛素抵抗,并独立于高血糖加重肾小球疾病。