Saini Heena, Hakeem Ifrah, Mukherjee Sudeshna, Chowdhury Shibasish, Chowdhury Rajdeep
Department of Biological Sciences, Pilani Campus, BITS, Pilani, Rajasthan 333031, India.
J Oncol. 2019 Jan 6;2019:6164807. doi: 10.1155/2019/6164807. eCollection 2019.
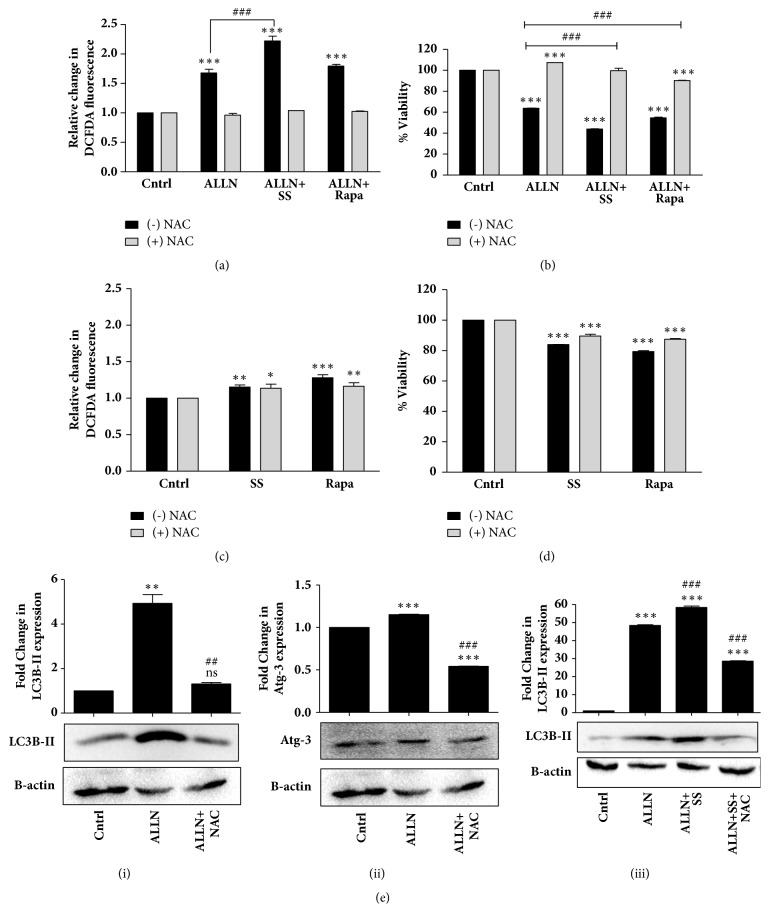
Mutations in p53, especially gain of function (GOF) mutations, are highly frequent in lung cancers and are known to facilitate tumor aggressiveness. Yet, the links between mutant GOF-p53 and lung cancers are not well established. In the present study, we set to examine how we can better sensitize resistant GOF-p53 lung cancer cells through modulation of cellular protein degradation machineries, proteasome and autophagy. H1299 p53 null lung cancer cells were stably transfected with R273H mutant GOF-p53 or wild-type (wt) p53 or empty vectors. The presence of R273H-P53 conferred the cancer cells with drug resistance not only against the widely used chemotherapeutic agents like cisplatin (CDDP) or 5-flurouracil (5-FU) but also against potent alternative modes of therapy like proteasomal inhibition. Therefore, there is an urgent need for new strategies that can overcome GOF-p53 induced drug resistance and prolong patient survival following failure of standard therapies. We observed that the proteasomal inhibitor, peptide aldehyde N-acetyl-leu-leu-norleucinal (commonly termed as ALLN), caused an activation of cellular homeostatic machinery, autophagy in R273H-P53 cells. Interestingly, inhibition of autophagy by chloroquine (CQ) alone or in combination with ALLN failed to induce enhanced cell death in the R273H-P53 cells; however, in contrast, an activation of autophagy by serum starvation or rapamycin increased sensitivity of cells to ALLN-induced cytotoxicity. An activated autophagy was associated with increased ROS and ERK signaling and an inhibition of either ROS or ERK signaling resulted in reduced cytotoxicity. Furthermore, inhibition of GOF-p53 was found to enhance autophagy resulting in increased cell death. Our findings provide novel insights pertaining to mechanisms by which a GOF-p53 harboring lung cancer cell is better sensitized, which can lead to the development of advanced therapy against resistant lung cancer cells.
p53突变,尤其是功能获得性(GOF)突变,在肺癌中非常常见,并且已知会促进肿瘤侵袭性。然而,突变型GOF-p53与肺癌之间的联系尚未完全确立。在本研究中,我们着手研究如何通过调节细胞蛋白质降解机制(蛋白酶体和自噬)来更好地使耐药性GOF-p53肺癌细胞敏感化。H1299 p53缺失的肺癌细胞用R273H突变型GOF-p53或野生型(wt)p53或空载体进行稳定转染。R273H-P53的存在赋予癌细胞不仅对广泛使用的化疗药物如顺铂(CDDP)或5-氟尿嘧啶(5-FU)耐药,而且对蛋白酶体抑制等有效的替代治疗模式也耐药。因此,迫切需要新的策略来克服GOF-p53诱导的耐药性,并在标准治疗失败后延长患者生存期。我们观察到蛋白酶体抑制剂肽醛N-乙酰-亮氨酸-亮氨酸-正亮氨酸(通常称为ALLN)在R273H-P53细胞中引起细胞稳态机制自噬的激活。有趣的是,单独使用氯喹(CQ)或与ALLN联合抑制自噬未能在R273H-P53细胞中诱导增强的细胞死亡;然而,相反的是,血清饥饿或雷帕霉素激活自噬增加了细胞对ALLN诱导的细胞毒性的敏感性。激活的自噬与活性氧(ROS)和细胞外信号调节激酶(ERK)信号增加相关,抑制ROS或ERK信号均导致细胞毒性降低。此外,发现抑制GOF-p53可增强自噬,从而导致细胞死亡增加。我们的研究结果为具有GOF-p53的肺癌细胞更好地敏感化的机制提供了新的见解,这可能导致针对耐药肺癌细胞的先进治疗方法的开发。