Computational Structural Biology Section, National Institutes of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD.
Max Planck Institute for Biophysics, Frankfurt am Main, Germany.
J Gen Physiol. 2019 Mar 4;151(3):381-394. doi: 10.1085/jgp.201812111. Epub 2019 Feb 6.
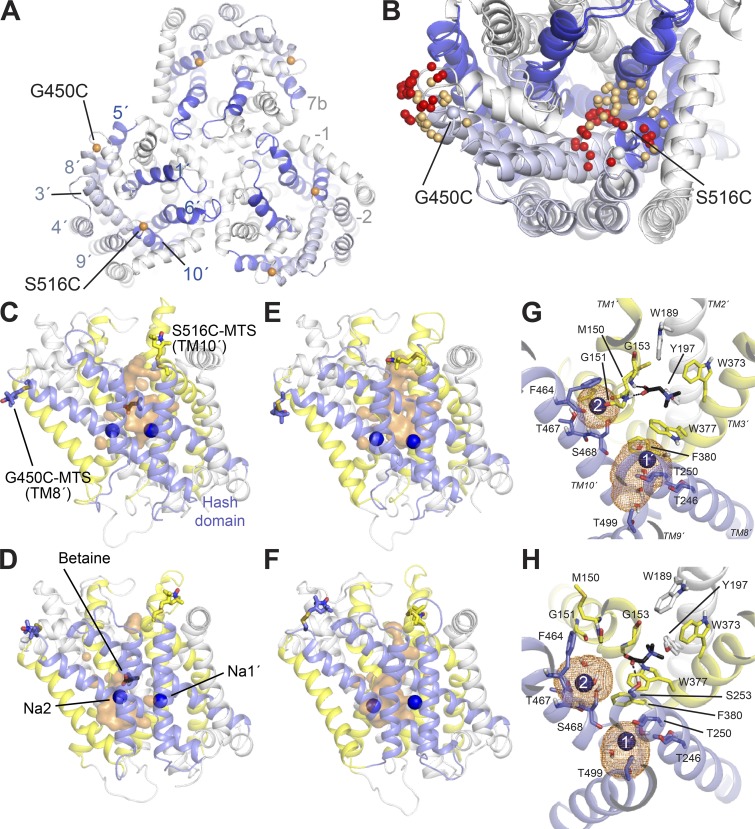
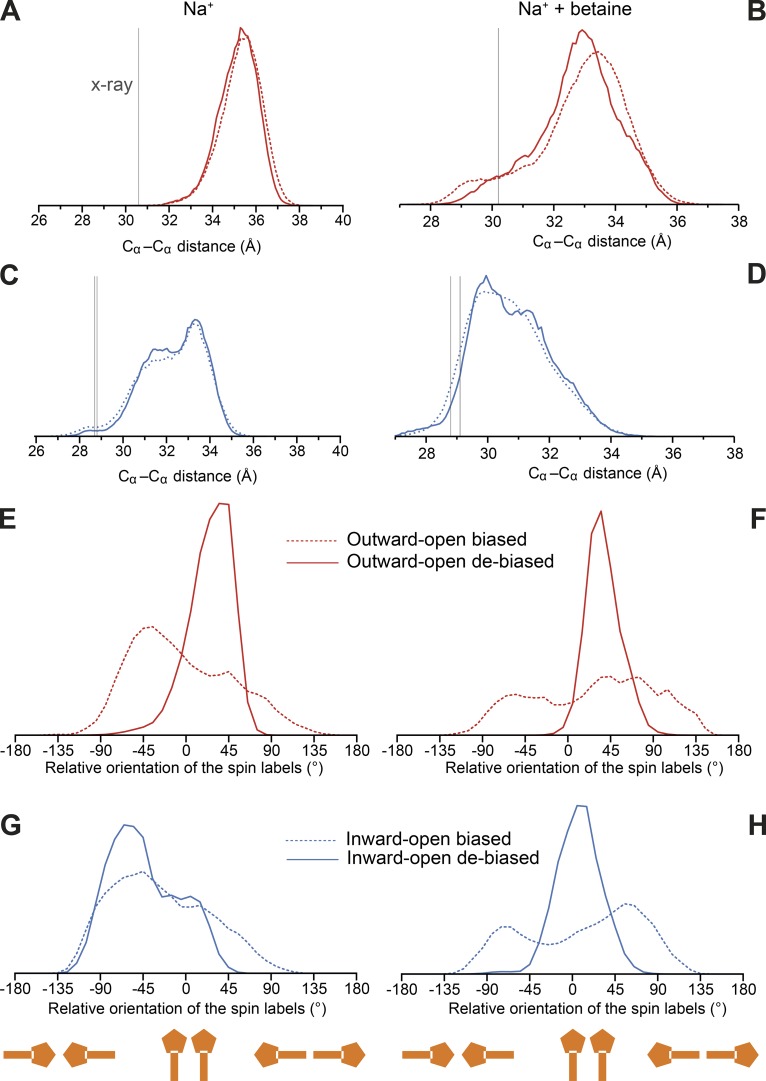
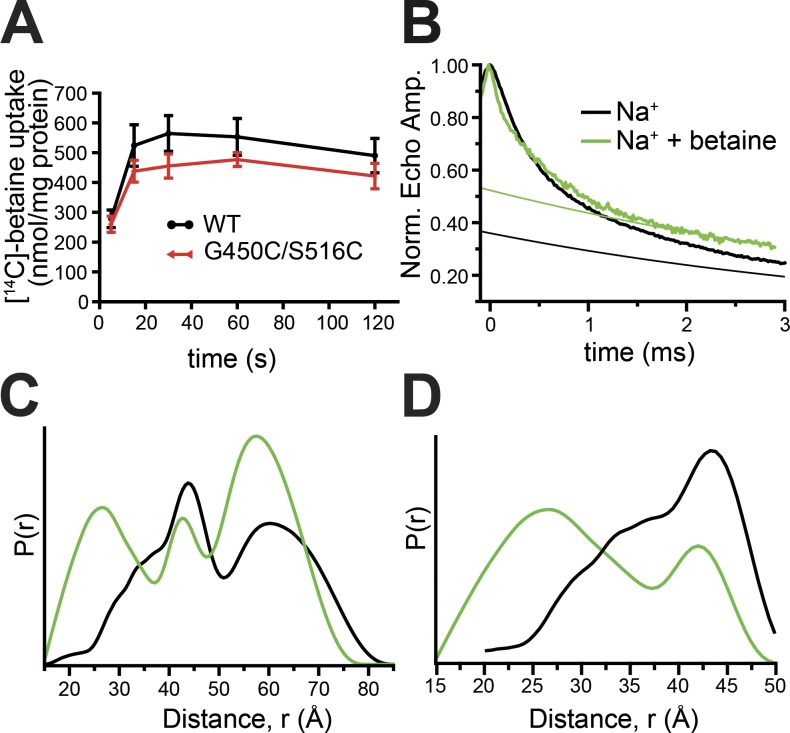
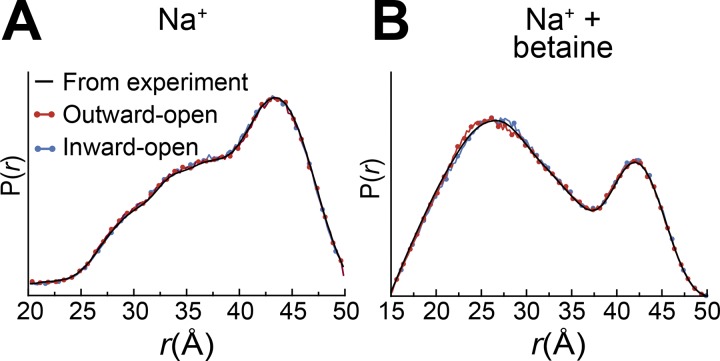
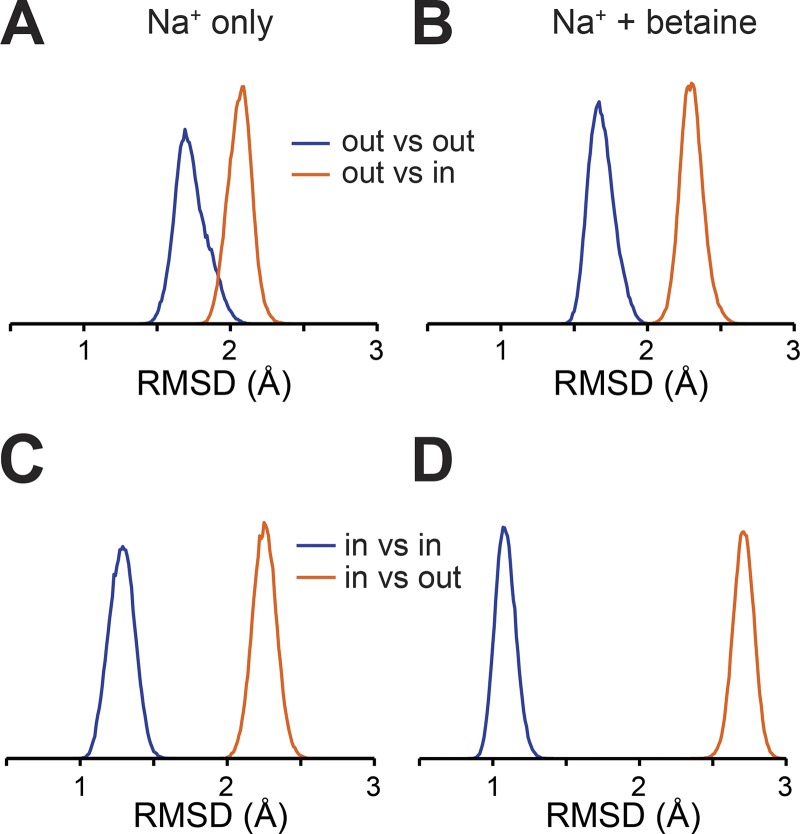
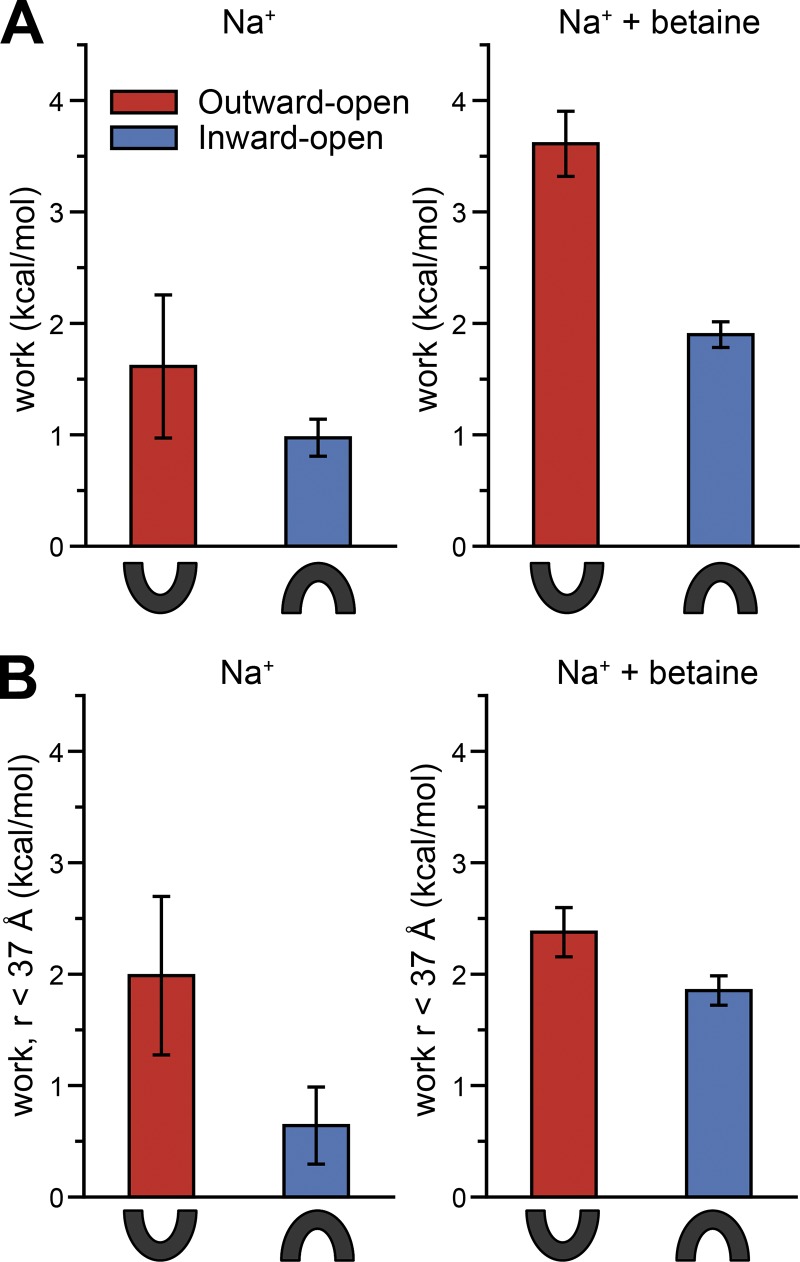
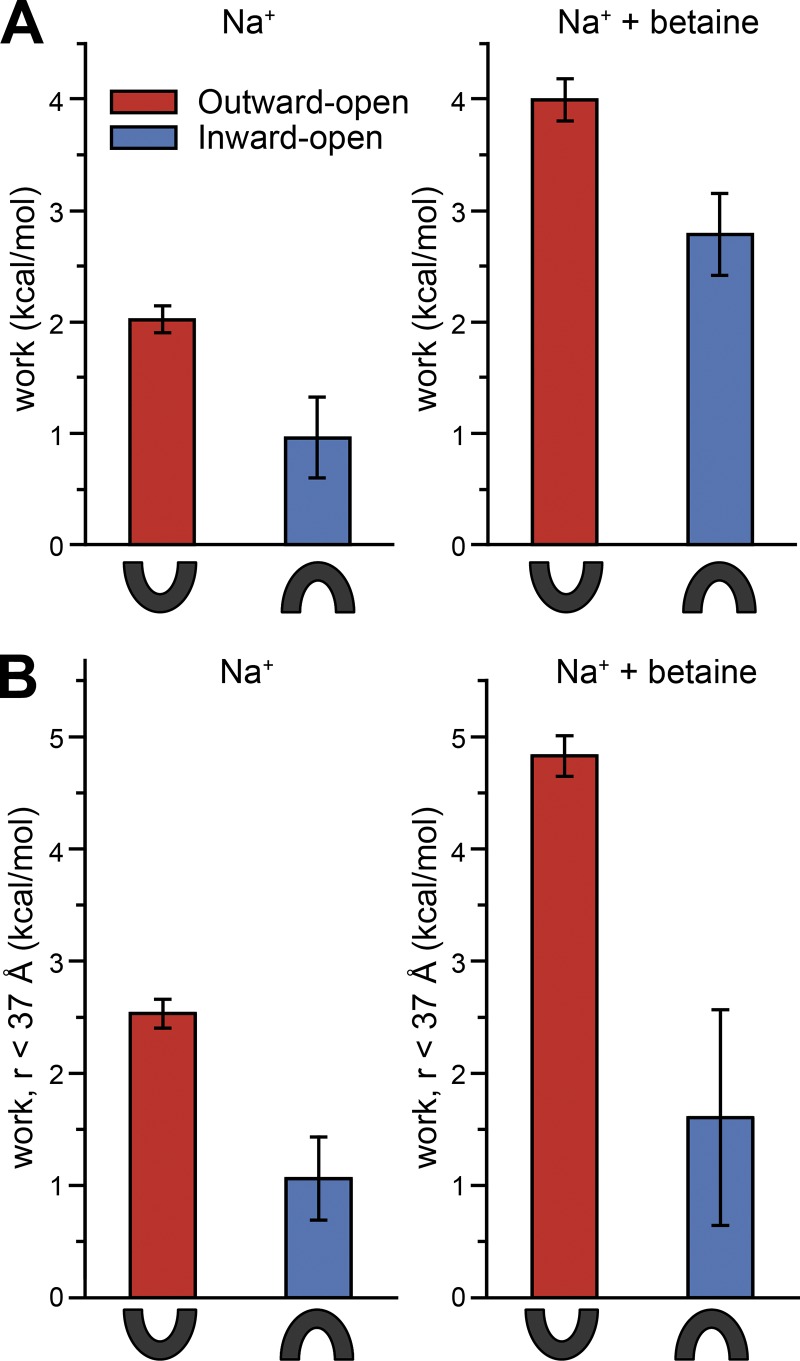
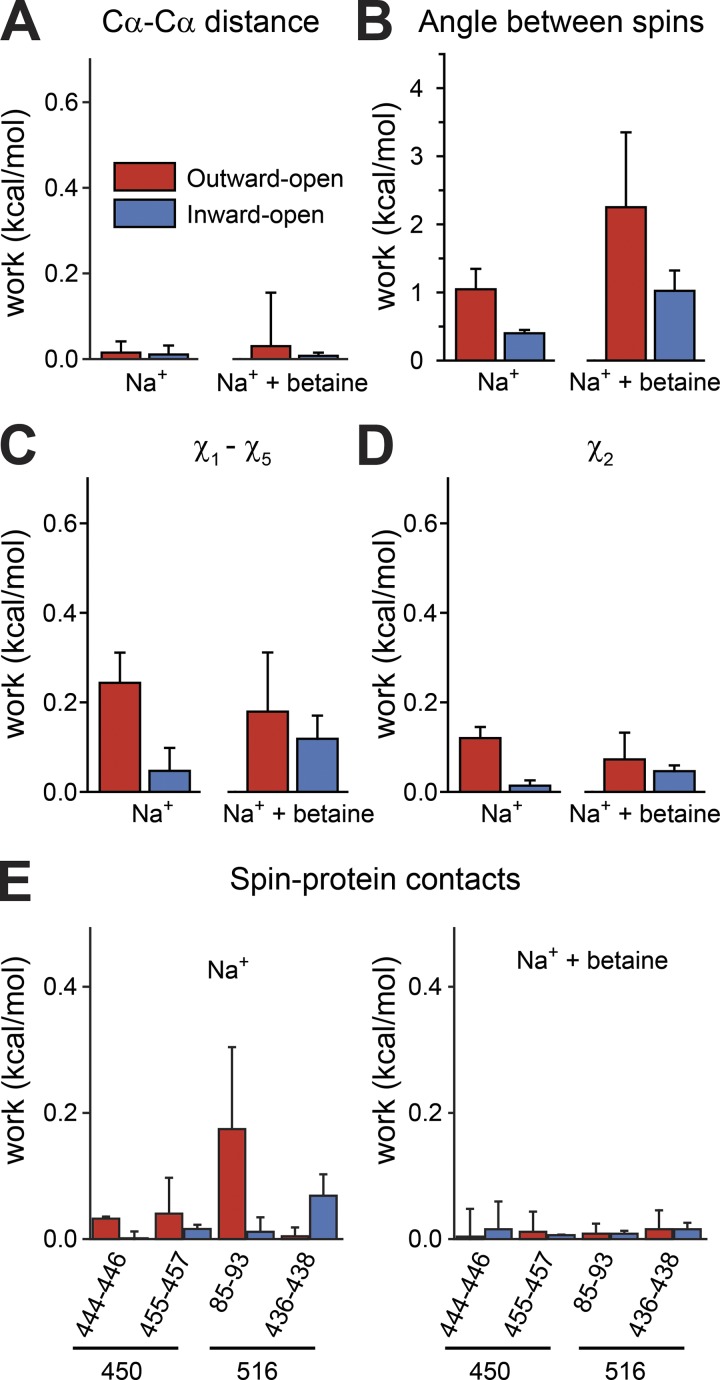
Mechanistic understanding of dynamic membrane proteins such as transporters, receptors, and channels requires accurate depictions of conformational ensembles, and the manner in which they interchange as a function of environmental factors including substrates, lipids, and inhibitors. Spectroscopic techniques such as electron spin resonance (ESR) pulsed electron-electron double resonance (PELDOR), also known as double electron-electron resonance (DEER), provide a complement to atomistic structures obtained from x-ray crystallography or cryo-EM, since spectroscopic data reflect an ensemble and can be measured in more native solvents, unperturbed by a crystal lattice. However, attempts to interpret DEER data are frequently stymied by discrepancies with the structural data, which may arise due to differences in conditions, the dynamics of the protein, or the flexibility of the attached paramagnetic spin labels. Recently, molecular simulation techniques such as EBMetaD have been developed that create a conformational ensemble matching an experimental distance distribution while applying the minimal possible bias. Moreover, it has been proposed that the work required during an EBMetaD simulation to match an experimentally determined distribution could be used as a metric with which to assign conformational states to a given measurement. Here, we demonstrate the application of this concept for a sodium-coupled transport protein, BetP. Because the probe, protein, and lipid bilayer are all represented in atomic detail, the different contributions to the work, such as the extent of protein backbone movements, can be separated. This work therefore illustrates how ranking simulations based on EBMetaD can help to bridge the gap between structural and biophysical data and thereby enhance our understanding of membrane protein conformational mechanisms.
机制理解动态膜蛋白,如转运蛋白、受体和通道,需要准确描述构象集合,以及它们作为环境因素(包括底物、脂质和抑制剂)的函数相互交换的方式。光谱技术,如电子自旋共振(ESR)脉冲电子-电子双共振(PELDOR),也称为双电子-电子共振(DEER),为从 X 射线晶体学或低温电子显微镜获得的原子结构提供了补充,因为光谱数据反映了一个集合,可以在更天然的溶剂中测量,不受晶格的干扰。然而,解释 DEER 数据的尝试经常因与结构数据的差异而受阻,这些差异可能是由于条件、蛋白质的动力学或附着的顺磁自旋标记的灵活性的差异引起的。最近,已经开发了分子模拟技术,如 EBMetaD,它可以创建一个构象集合,该集合与实验距离分布匹配,同时应用最小可能的偏差。此外,有人提出,在 EBMetaD 模拟中匹配实验确定的分布所需的工作可以用作指标,将构象状态分配给给定的测量。在这里,我们展示了这个概念在钠耦合转运蛋白 BetP 中的应用。由于探针、蛋白质和脂质双层都以原子细节表示,因此可以分离工作的不同贡献,例如蛋白质骨架运动的程度。因此,这项工作说明了如何根据 EBMetaD 对模拟进行排名,可以帮助弥合结构和生物物理数据之间的差距,从而增强我们对膜蛋白构象机制的理解。