Reference center of Inherited Metabolic disorder, CERLYMM, Département de Pédiatrie, HCL Hopital Femme Mère Enfant, 59 Boulevard Pinel, 69677, Bron cedex, France.
Paediatric Orthopaedic Surgery Department, Lorraine University Hospital Centre, Children's Hospital, Vandoeuvre lès Nancy, France.
Eur J Pediatr. 2019 Apr;178(4):593-603. doi: 10.1007/s00431-019-03330-x. Epub 2019 Feb 11.
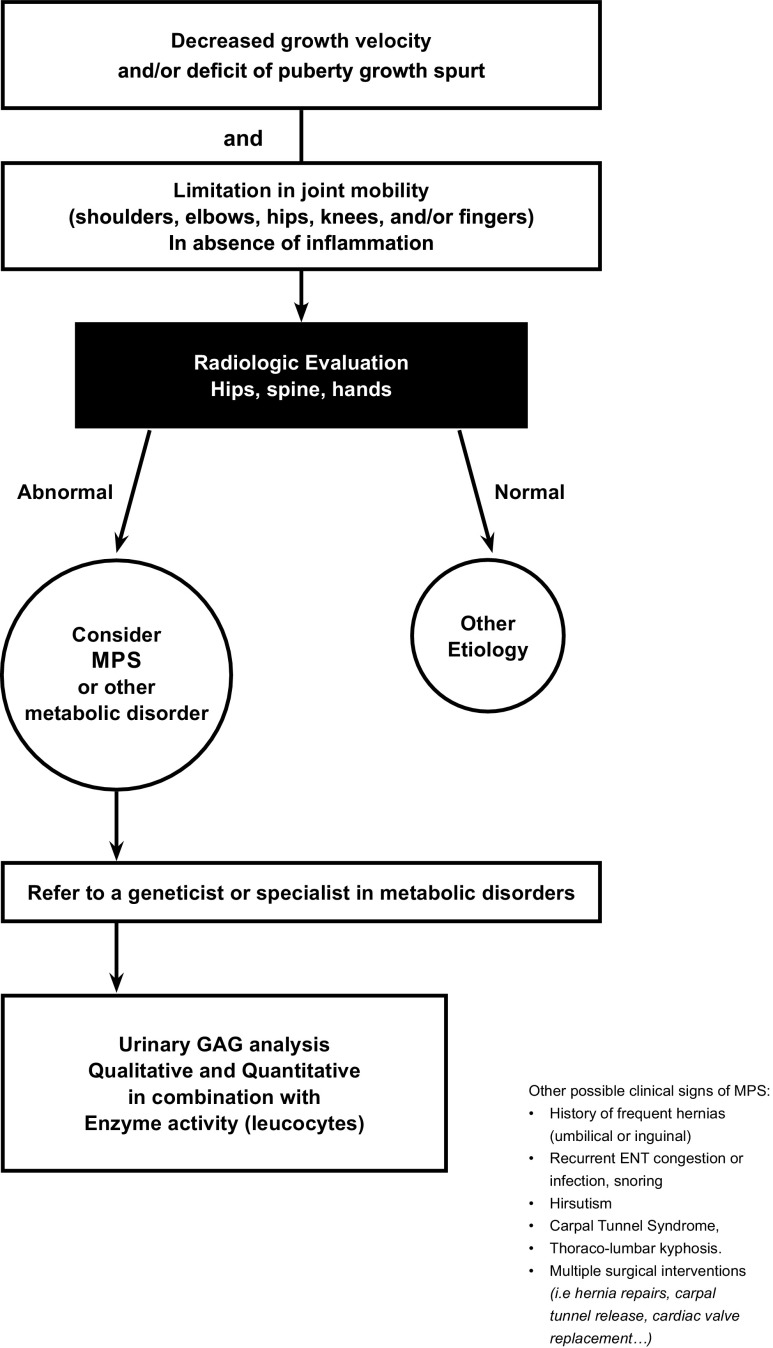
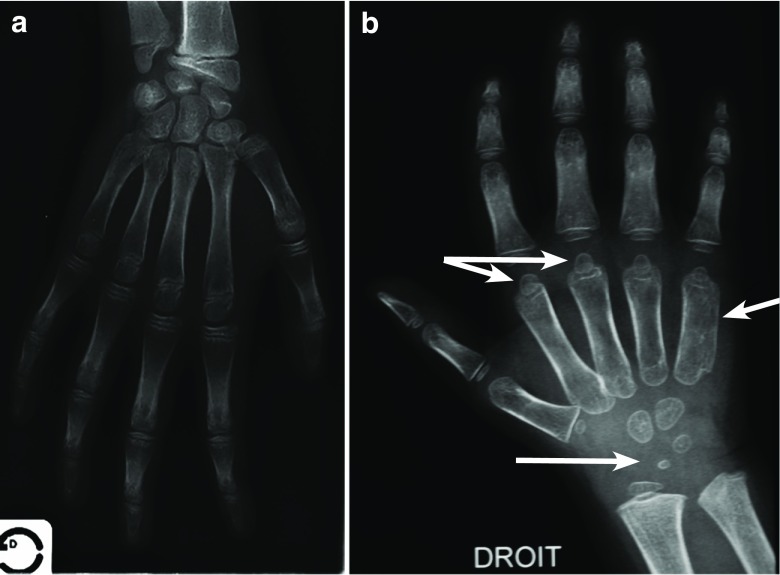
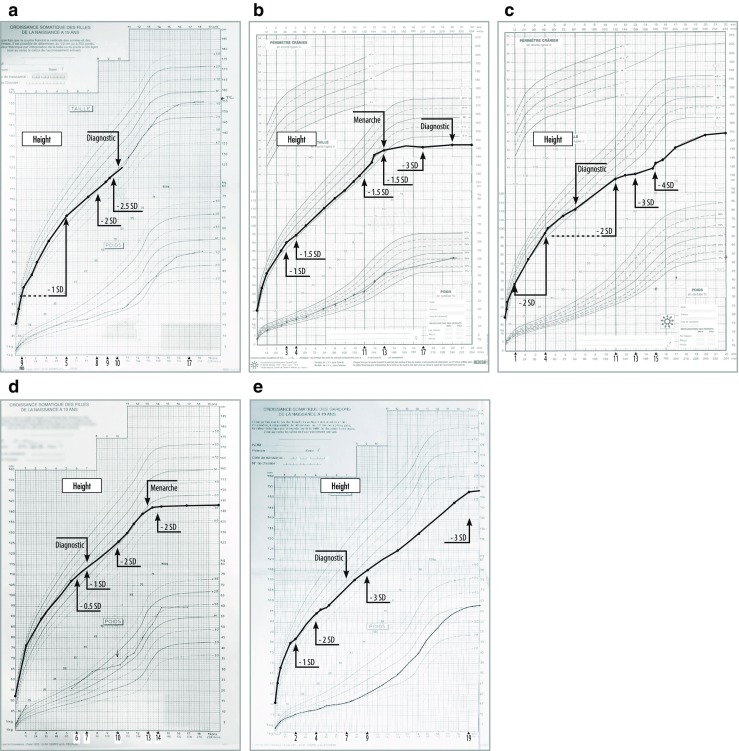
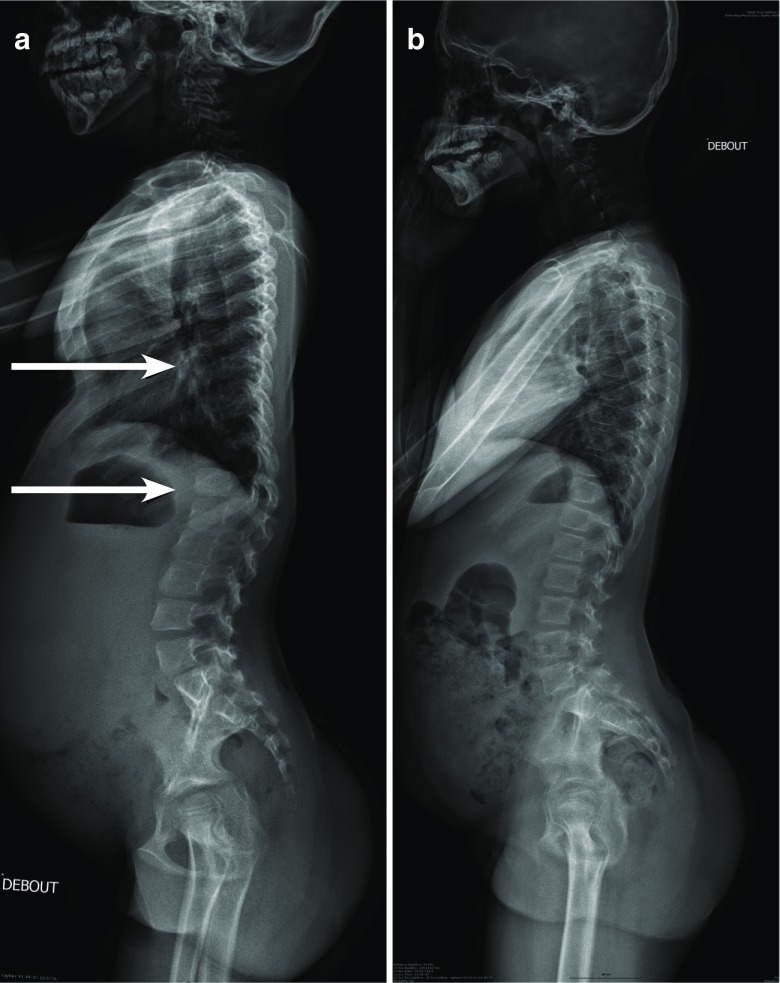
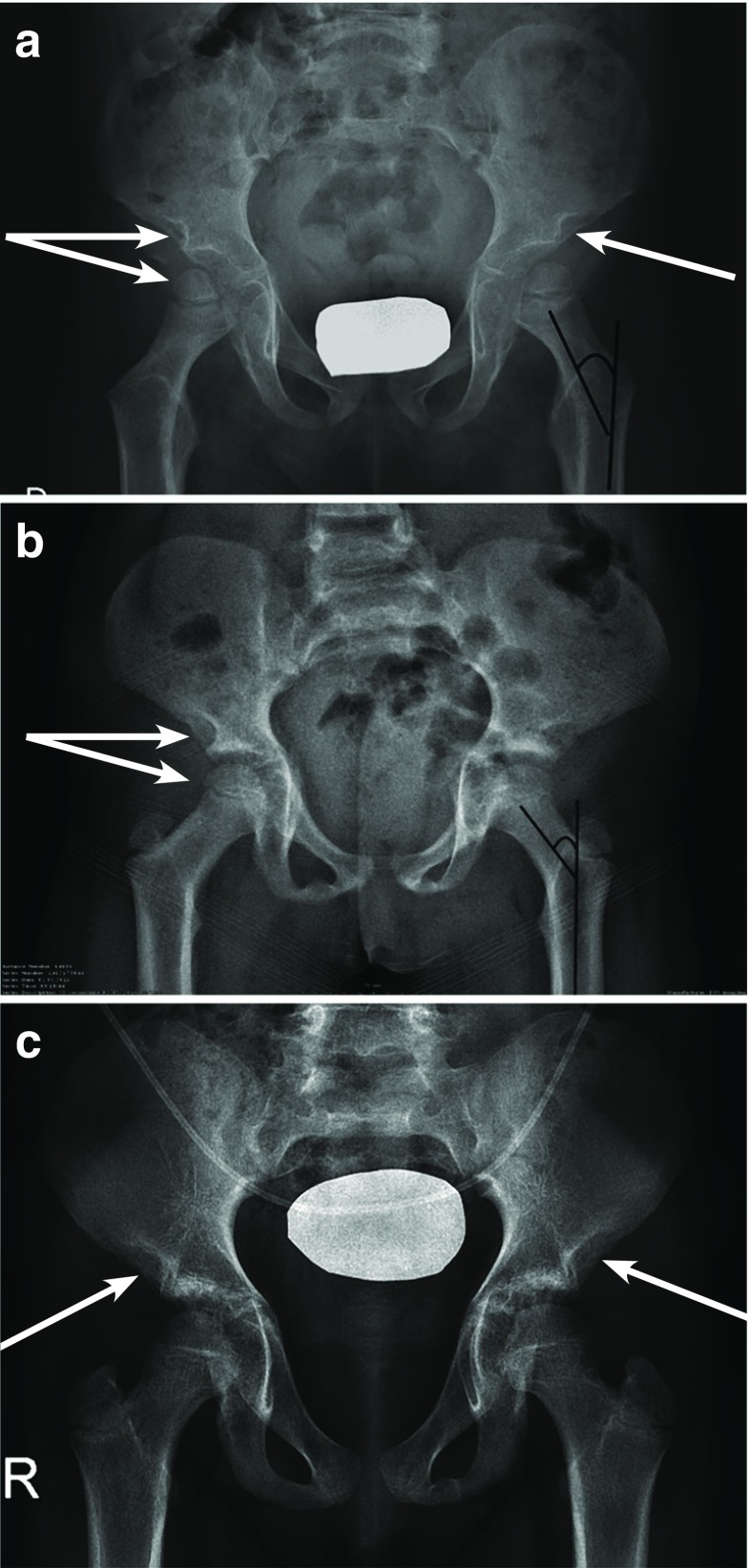
Growth impairment together with bone and joint involvement is common to most patients with mucopolysaccharidosis (MPS) disorders. The genetic basis for these metabolic disorders involves various enzyme deficiencies responsible for the catabolism of glycosaminoglycans (GAGs). The incomplete degradation and subsequent accumulation of GAGs result in progressive tissue damage throughout the body. Bone ossification is particularly affected, with the consequent onset of dysostosis multiplex which is the underlying cause of short stature. Joint manifestations, whether joint contractures (MPS I, II, VI, VII) or hyperlaxity (MPS IV), affect fine motor skills and quality of life. Subtle decreases in growth velocity can begin as early as 2-4 years of age. Pediatricians are in the front line to recognize or suspect MPS. However, given the rarity of the disorders and variable ages of symptom onset depending on disease severity, recognition and diagnostic delays remain a challenge, especially for the attenuated forms. Prompt diagnosis and treatment can prevent irreversible disease outcomes.Conclusion: We present a diagnostic algorithm based on growth velocity decline and bone and joint involvement designed to help pediatricians recognize early manifestations of attenuated forms of MPS. We illustrate the paper with examples of abnormal growth curves and subtle radiographic nuances. What is Known: • As mucopolysaccharidoses (MPSs) are rare genetic disorders infrequently seen in clinical practice, there can be a lag between symptom onset and diagnosis, especially of attenuated forms of the disease. • This highlights the need for increased disease awareness to recognize early clinical signs and subsequently initiate early treatment to improve outcomes (normal height potential) and possibly prevent or delay the development of irreversible disease manifestations. What is New: • Growth impairment co-presenting with limited range of joint motion and radiographic anomalies in children should raise suspicions of possible attenuated MPS (AMPS). • Experts present a diagnostic algorithm with detailed focus on the decline in growth velocity, delayed puberty and limitation in joint mobility seen in children with AMPS, to shorten time-to-diagnosis and treatment and potentially improve patient outcome.
生长障碍以及骨骼和关节受累是大多数黏多糖贮积症(MPS)患者的共同特征。这些代谢紊乱的遗传基础涉及各种负责糖胺聚糖(GAG)分解代谢的酶缺乏。GAG 不完全降解和随后的积累导致全身组织逐渐受损。骨骼的骨化特别受到影响,随之而来的是多发性发育不良的发生,这是身材矮小的根本原因。关节表现,无论是关节挛缩(MPS I、II、VI、VII)还是过度松弛(MPS IV),都会影响精细运动技能和生活质量。生长速度的细微下降可能早在 2-4 岁时就开始出现。儿科医生处于识别或怀疑 MPS 的第一线。然而,由于这些疾病的罕见性以及症状发作的年龄因疾病严重程度而异,因此识别和诊断延迟仍然是一个挑战,尤其是对于轻度形式。及时诊断和治疗可以预防不可逆转的疾病结局。结论:我们提出了一种基于生长速度下降和骨骼关节受累的诊断算法,旨在帮助儿科医生识别 MPS 轻度形式的早期表现。我们用异常生长曲线和细微的放射学细微差别来说明本文。已知的是:• 由于黏多糖贮积症(MPS)是罕见的遗传疾病,在临床实践中很少见,因此从症状发作到诊断可能存在时间滞后,尤其是对于疾病的轻度形式。• 这强调了需要提高疾病意识,以识别早期临床体征,随后尽早开始治疗,以改善结果(正常身高潜力),并可能预防或延迟不可逆疾病表现的发展。新的是:• 儿童出现生长障碍,伴有关节活动范围有限和放射学异常,应怀疑可能存在轻度 MPS(AMPS)。• 专家提出了一种诊断算法,详细重点关注 AMPS 儿童的生长速度下降、青春期延迟和关节活动受限,以缩短诊断和治疗时间,并有可能改善患者的预后。