Yuan Yibing, Cairns Jill E, Babu Raman, Gowda Manje, Makumbi Dan, Magorokosho Cosmos, Zhang Ao, Liu Yubo, Wang Nan, Hao Zhuanfang, San Vicente Felix, Olsen Michael S, Prasanna Boddupalli M, Lu Yanli, Zhang Xuecai
Maize Research Institute, Sichuan Agricultural University, Wenjiang, China.
Key Laboratory of Biology and Genetic Improvement of Maize in Southwest Region, Ministry of Agriculture, Chengdu, China.
Front Plant Sci. 2019 Jan 30;9:1919. doi: 10.3389/fpls.2018.01919. eCollection 2018.
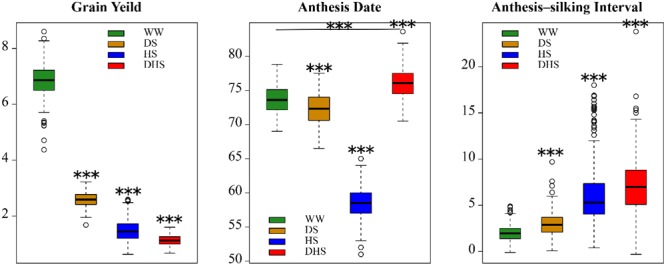
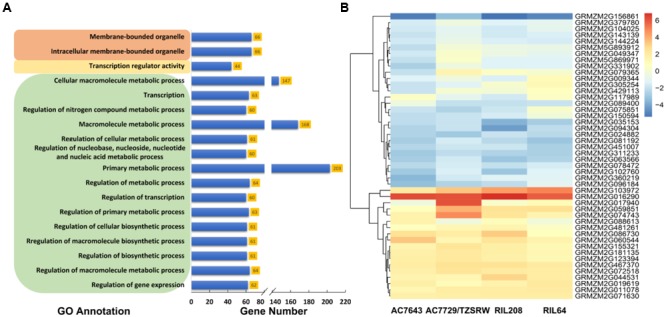
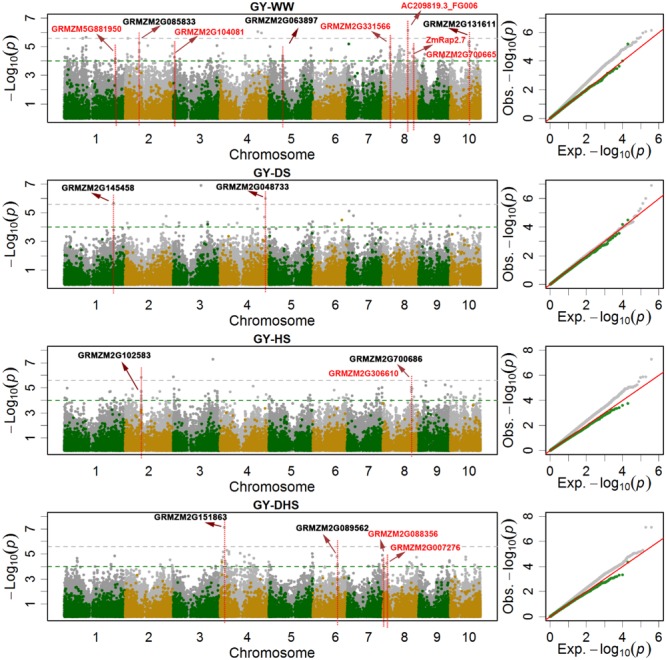
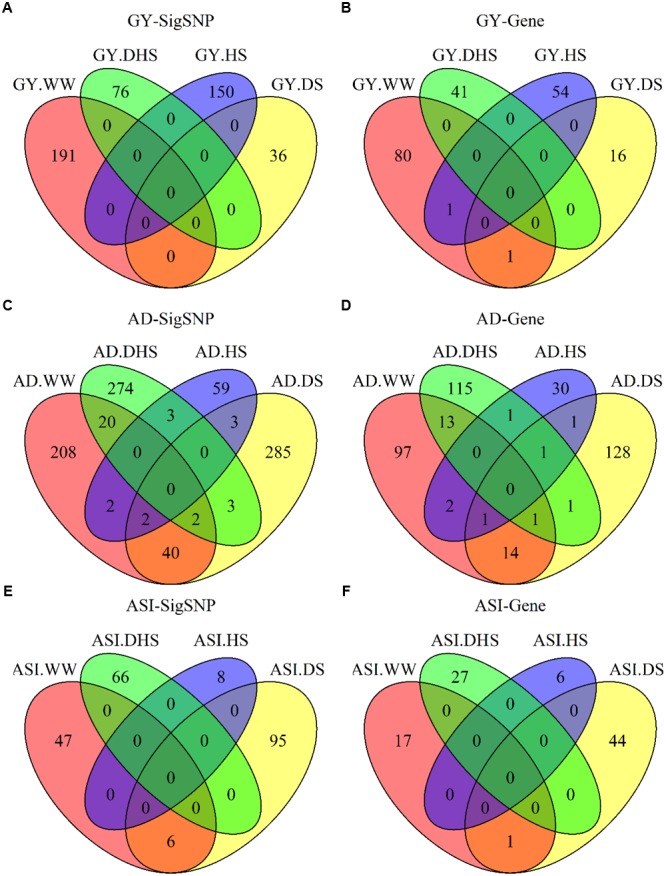
Drought stress (DS) is a major constraint to maize yield production. Heat stress (HS) alone and in combination with DS are likely to become the increasing constraints. Association mapping and genomic prediction (GP) analyses were conducted in a collection of 300 tropical and subtropical maize inbred lines to reveal the genetic architecture of grain yield and flowering time under well-watered (WW), DS, HS, and combined DS and HS conditions. Out of the 381,165 genotyping-by-sequencing SNPs, 1549 SNPs were significantly associated with all the 12 trait-environment combinations, the average PVE (phenotypic variation explained) by these SNPs was 4.33%, and 541 of them had a PVE value greater than 5%. These significant associations were clustered into 446 genomic regions with a window size of 20 Mb per region, and 673 candidate genes containing the significantly associated SNPs were identified. In addition, 33 hotspots were identified for 12 trait-environment combinations and most were located on chromosomes 1 and 8. Compared with single SNP-based association mapping, the haplotype-based associated mapping detected fewer number of significant associations and candidate genes with higher PVE values. All the 688 candidate genes were enriched into 15 gene ontology terms, and 46 candidate genes showed significant differential expression under the WW and DS conditions. Association mapping results identified few overlapped significant markers and candidate genes for the same traits evaluated under different managements, indicating the genetic divergence between the individual stress tolerance and the combined drought and HS tolerance. The GP accuracies obtained from the marker-trait associated SNPs were relatively higher than those obtained from the genome-wide SNPs for most of the target traits. The genetic architecture information of the grain yield and flowering time revealed in this study, and the genomic regions identified for the different trait-environment combinations are useful in accelerating the efforts on rapid development of the stress-tolerant maize germplasm through marker-assisted selection and/or genomic selection.
干旱胁迫(DS)是玉米产量的主要限制因素。单独的热胁迫(HS)以及与DS相结合的情况可能会成为日益增加的限制因素。对300个热带和亚热带玉米自交系进行了关联分析和基因组预测(GP)分析,以揭示在水分充足(WW)、DS、HS以及DS和HS组合条件下籽粒产量和开花时间的遗传结构。在通过测序进行基因分型的381,165个单核苷酸多态性(SNP)中,有1549个SNP与所有12种性状-环境组合显著相关,这些SNP解释的平均表型变异(PVE)为4.33%,其中541个的PVE值大于5%。这些显著关联被聚类到446个基因组区域,每个区域的窗口大小为20兆碱基对(Mb),并鉴定出673个包含显著相关SNP的候选基因。此外,针对12种性状-环境组合鉴定出33个热点区域,大多数位于第1和第8号染色体上。与基于单个SNP的关联分析相比,基于单倍型的关联分析检测到的显著关联和具有较高PVE值的候选基因数量更少。所有688个候选基因被富集到15个基因本体论术语中,46个候选基因在WW和DS条件下表现出显著的差异表达。关联分析结果表明,在不同管理条件下评估的相同性状,几乎没有重叠的显著标记和候选基因,这表明个体胁迫耐受性与干旱和HS组合耐受性之间存在遗传差异。对于大多数目标性状,从标记-性状相关SNP获得的GP准确性相对高于从全基因组SNP获得的准确性。本研究揭示的籽粒产量和开花时间的遗传结构信息,以及针对不同性状-环境组合鉴定出的基因组区域有助于通过标记辅助选择和/或基因组选择加速耐胁迫玉米种质的快速开发。