Mongia Aanchal, Sengupta Debarka, Majumdar Angshul
Department of Computer Science and Engineering, Indraprastha Institute of Information Technology Delhi, New Delhi, India.
Center for Computational Biology, Indraprastha Institute of Information Technology Delhi, New Delhi, India.
Front Genet. 2019 Jan 29;10:9. doi: 10.3389/fgene.2019.00009. eCollection 2019.
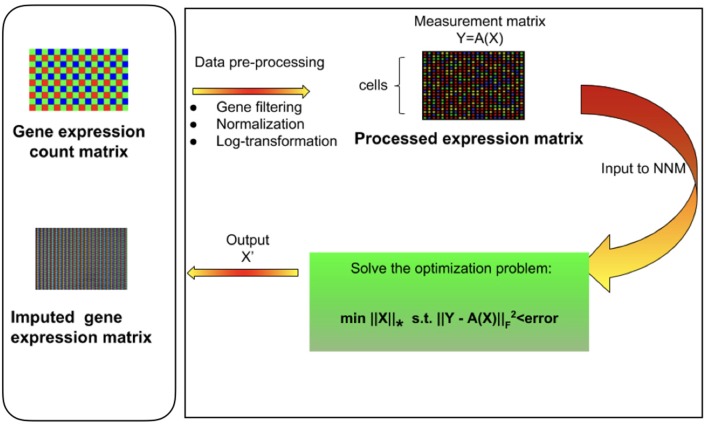
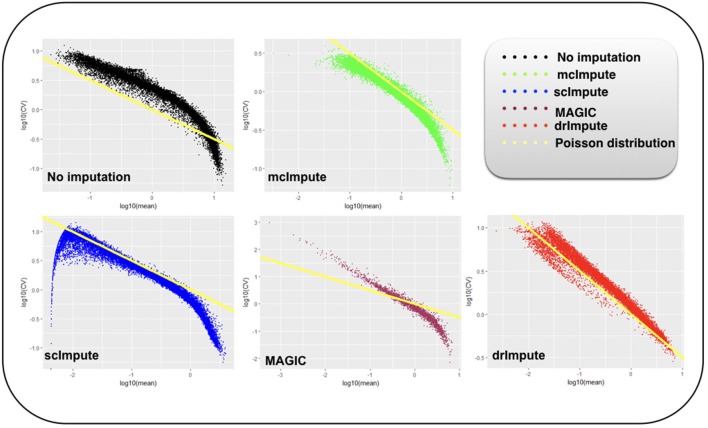
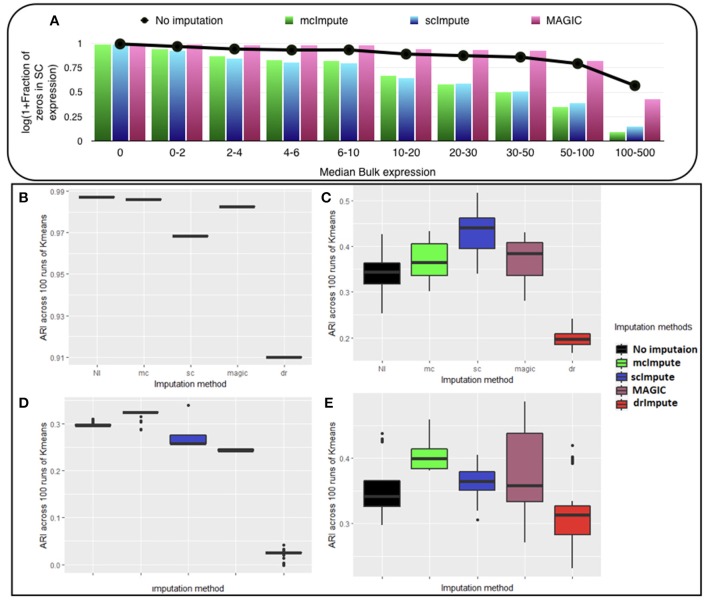
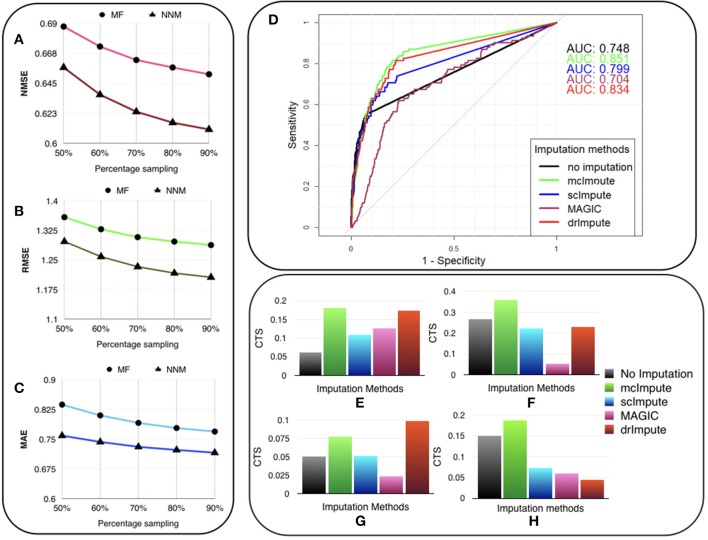
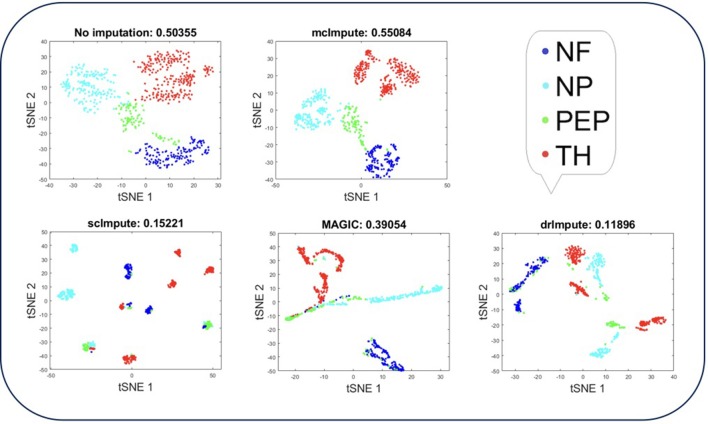
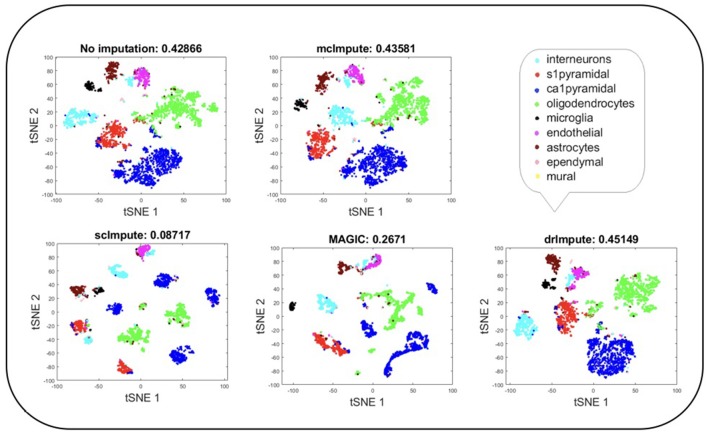
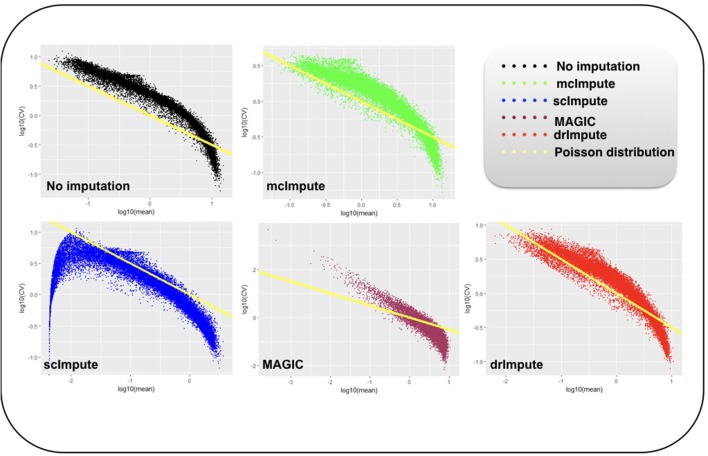
Single-cell RNA sequencing has been proved to be revolutionary for its potential of zooming into complex biological systems. Genome-wide expression analysis at single-cell resolution provides a window into dynamics of cellular phenotypes. This facilitates the characterization of transcriptional heterogeneity in normal and diseased tissues under various conditions. It also sheds light on the development or emergence of specific cell populations and phenotypes. However, owing to the paucity of input RNA, a typical single cell RNA sequencing data features a high number of dropout events where transcripts fail to get amplified. We introduce mcImpute, a low-rank matrix completion based technique to impute dropouts in single cell expression data. On a number of real datasets, application of mcImpute yields significant improvements in the separation of true zeros from dropouts, cell-clustering, differential expression analysis, cell type separability, the performance of dimensionality reduction techniques for cell visualization, and gene distribution. https://github.com/aanchalMongia/McImpute_scRNAseq.
单细胞RNA测序因其深入研究复杂生物系统的潜力而被证明具有革命性。单细胞分辨率下的全基因组表达分析为细胞表型动态提供了一个窗口。这有助于表征各种条件下正常组织和患病组织中的转录异质性。它还揭示了特定细胞群体和表型的发育或出现情况。然而,由于输入RNA的稀缺,典型的单细胞RNA测序数据具有大量的缺失事件,即转录本未能得到扩增。我们引入了mcImpute,一种基于低秩矩阵补全的技术,用于估算单细胞表达数据中的缺失值。在多个真实数据集上,应用mcImpute在区分真正的零值与缺失值、细胞聚类、差异表达分析、细胞类型可分离性、用于细胞可视化的降维技术性能以及基因分布方面都有显著改进。https://github.com/aanchalMongia/McImpute_scRNAseq 。