Wang Bruce, Rudnick Sean, Cengia Brent, Bonkovsky Herbert L
Division of Gastroenterology, Department of Medicine University of California San Francisco San Francisco CA.
Section of Gastroenterology and Hepatology, Department of Internal Medicine Wake Forest University School of Medicine Winston-Salem NC.
Hepatol Commun. 2018 Dec 20;3(2):193-206. doi: 10.1002/hep4.1297. eCollection 2019 Feb.
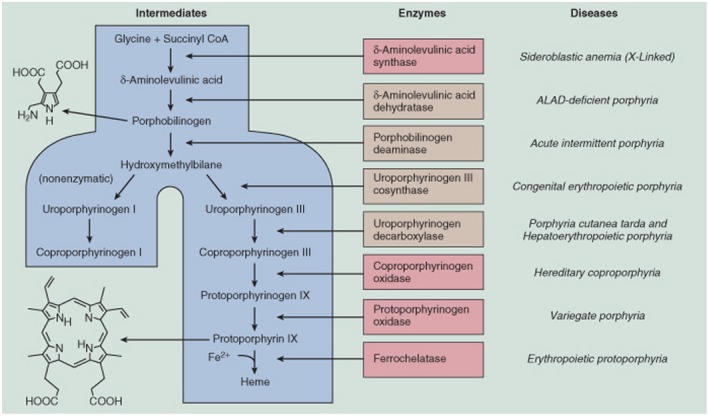
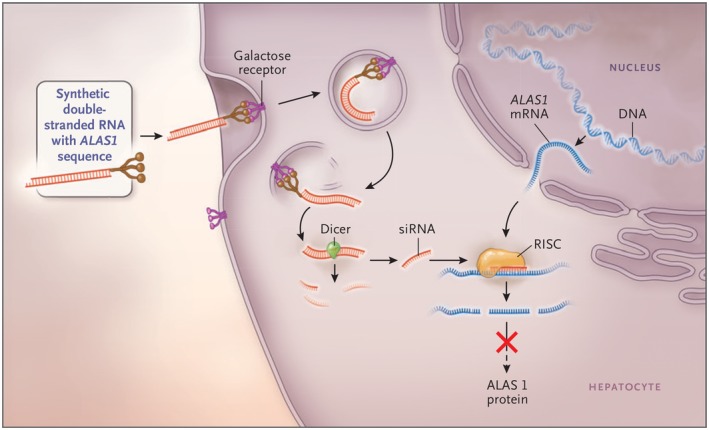
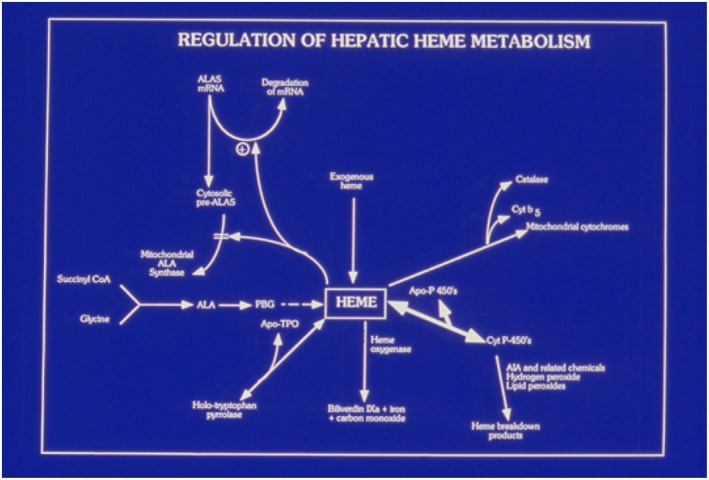
The acute hepatic porphyrias (AHPs) are a group of four inherited diseases of heme biosynthesis that present with episodic, acute neurovisceral symptoms. The four types are 5-aminolevulinic acid (ALA) dehydratase deficiency porphyria, acute intermittent porphyria, hereditary coproporphyria, and variegate porphyria. Their diagnoses are often missed or delayed because the clinical symptoms mimic other more common disorders. Recent results indicate that acute intermittent porphyria, the most severe of the more common types of AHP, is more prevalent than previously thought, occurring in about 1 in 1600 Caucasians, but with low clinical penetrance (approximately 2%-3%). Here we provide an updated review of relevant literature and discuss recent and emerging advances in treatment of these disorders. Symptomatic attacks occur primarily in females between 14 and 45 years of age. AHP is diagnosed by finding significantly elevated levels of porphyrin precursors ALA and porphobilinogen in urine. Acute attacks should be treated promptly with intravenous heme therapy to avoid the development of potentially irreversible neurologic sequelae. All patients should be counseled about avoiding potential triggers for acute attacks and monitored regularly for the development of long-term complications. Their first-degree relatives should undergo targeted gene testing. Patients who suffer recurrent acute attacks can be particularly challenging to manage. Approximately 20% of patients with recurrent symptoms develop chronic and ongoing pain and other symptoms. We discuss newer treatment options in development, including small interfering RNA, to down-regulate ALA synthase-1 and/or wild-type messenger RNA of defective genes delivered selectively to hepatocytes for these patients. We expect that the newer treatments will diminish and perhaps obviate the need for liver transplantation as treatment of these inborn metabolic disorders.
急性肝卟啉病(AHP)是一组四种遗传性血红素生物合成疾病,表现为发作性急性神经内脏症状。这四种类型分别是5-氨基酮戊酸(ALA)脱水酶缺乏性卟啉病、急性间歇性卟啉病、遗传性粪卟啉病和混合型卟啉病。由于临床症状与其他更常见疾病相似,其诊断常常被漏诊或延误。最近的结果表明,急性间歇性卟啉病是AHP较常见类型中最严重的一种,其患病率比以前认为的更高,约每1600名白种人中就有1例,但临床外显率较低(约2%-3%)。在此,我们提供相关文献的最新综述,并讨论这些疾病治疗方面的最新进展和新出现的进展。症状性发作主要发生在14至45岁的女性中。通过检测尿液中卟啉前体ALA和胆色素原水平显著升高来诊断AHP。急性发作应立即用静脉注射血红素疗法进行治疗,以避免潜在的不可逆神经后遗症的发生。应建议所有患者避免急性发作的潜在诱因,并定期监测长期并发症的发生。他们的一级亲属应接受靶向基因检测。患有复发性急性发作的患者管理起来可能特别具有挑战性。约20%有复发性症状的患者会出现慢性持续性疼痛和其他症状。我们讨论了正在研发的新治疗选择,包括小干扰RNA,以下调ALA合酶-1和/或缺陷基因的野生型信使RNA,并将其选择性地递送至这些患者的肝细胞。我们预计,新的治疗方法将减少甚至可能消除对肝移植作为这些先天性代谢疾病治疗手段的需求。