Institute of Cellular Biochemistry, University Medical Center Goettingen, Goettingen, Germany.
International Max-Planck Research School in Neuroscience, Goettingen, Germany.
Elife. 2019 Feb 18;8:e39598. doi: 10.7554/eLife.39598.
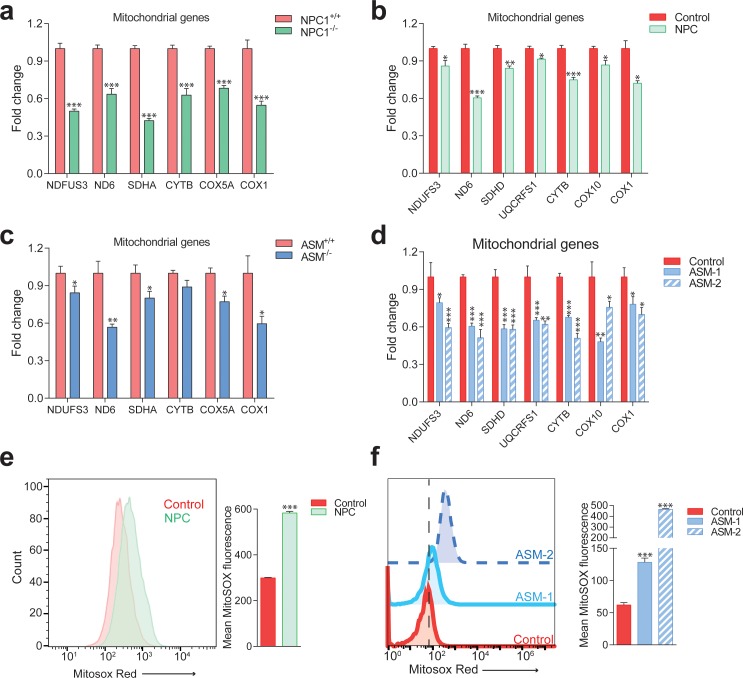
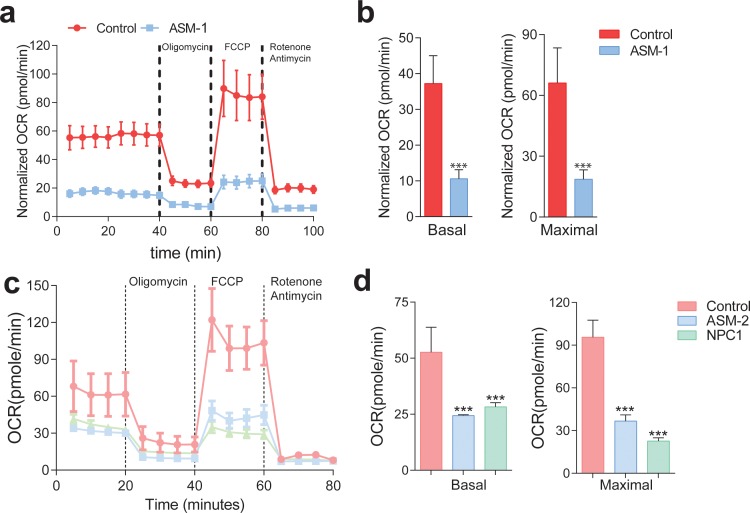
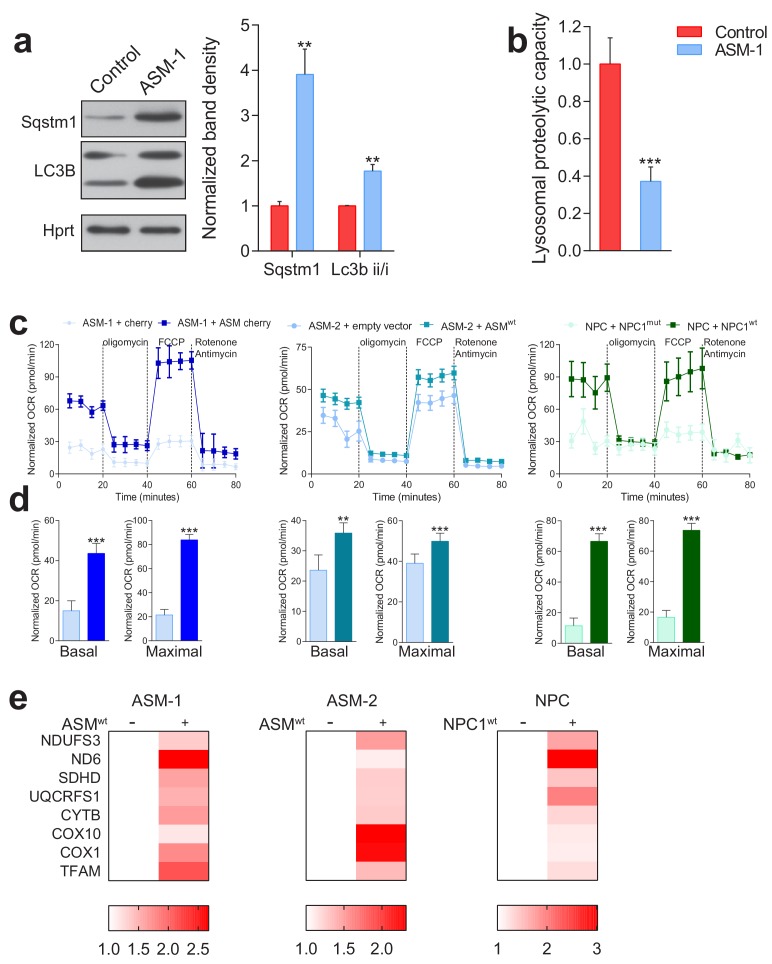
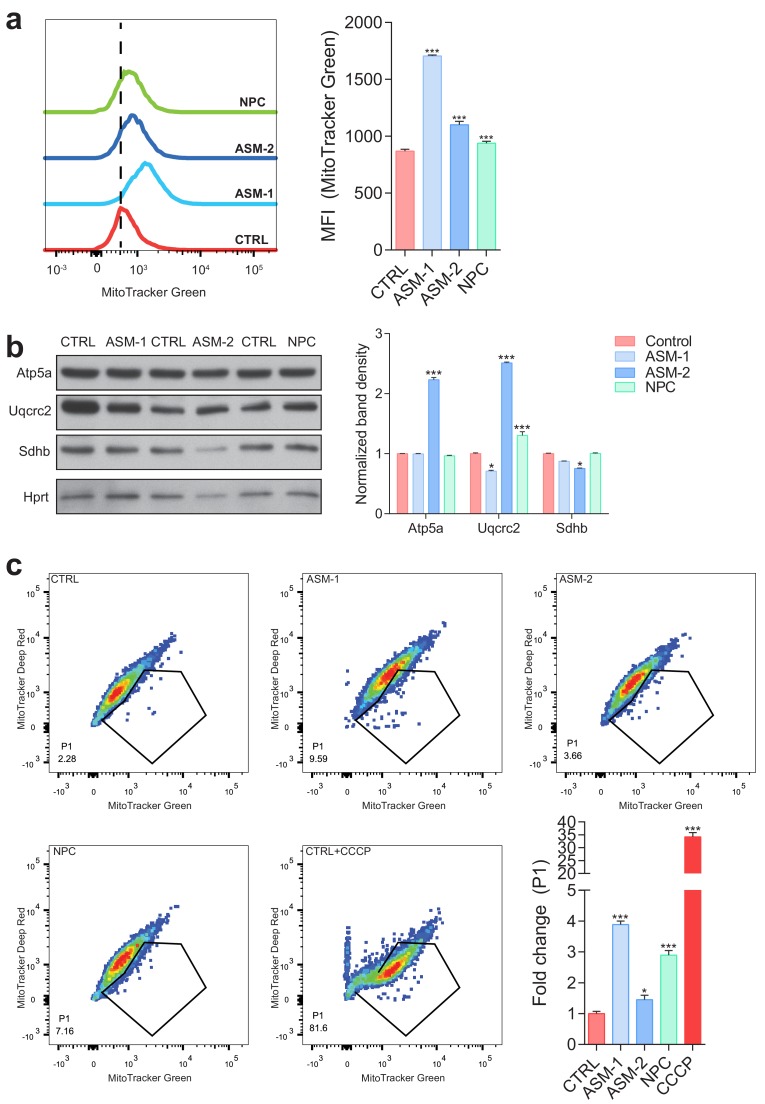
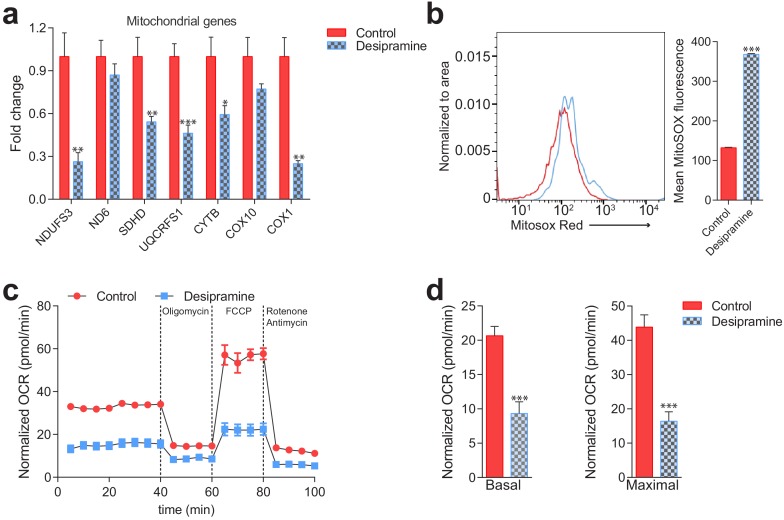
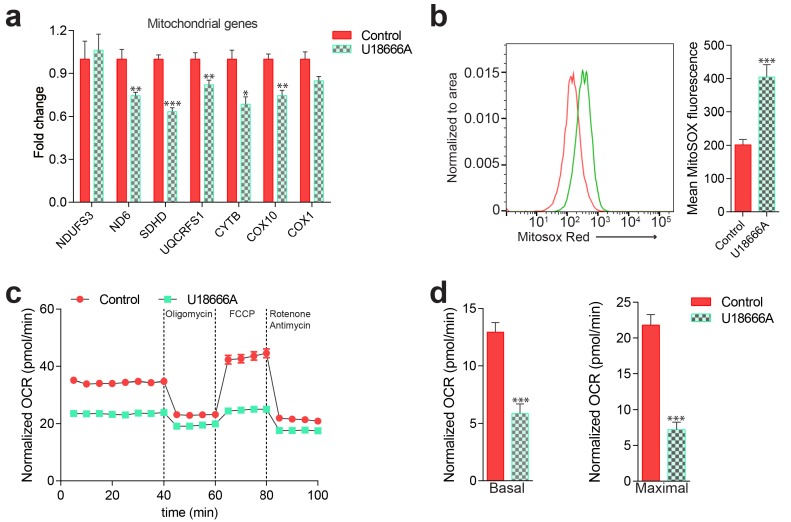
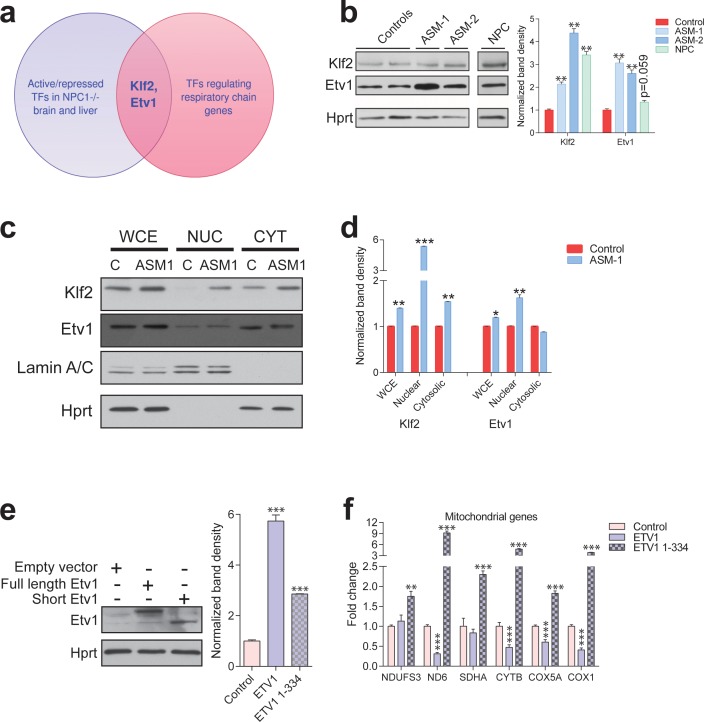
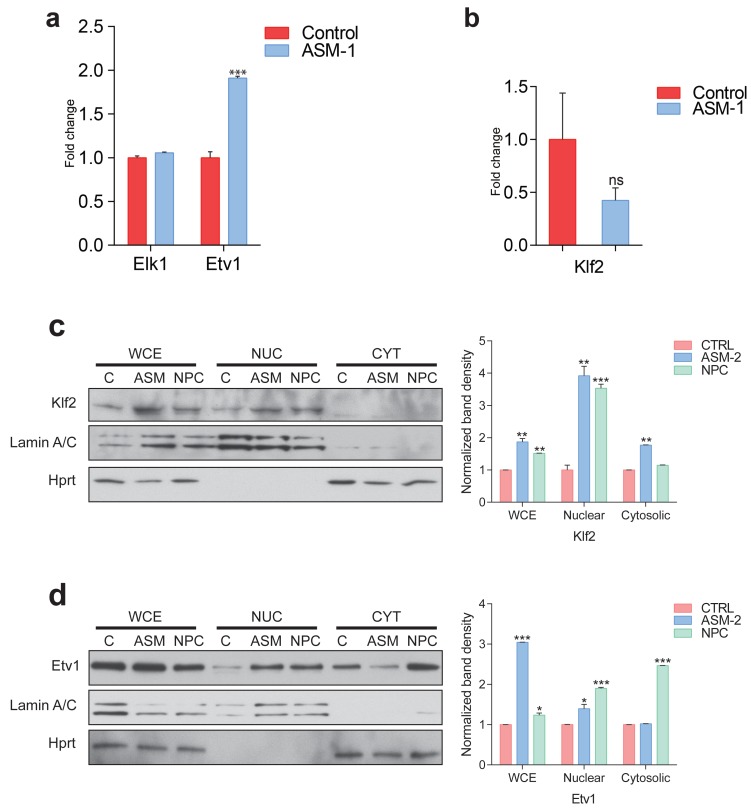
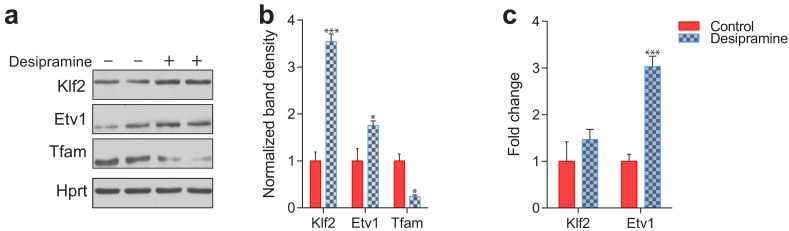
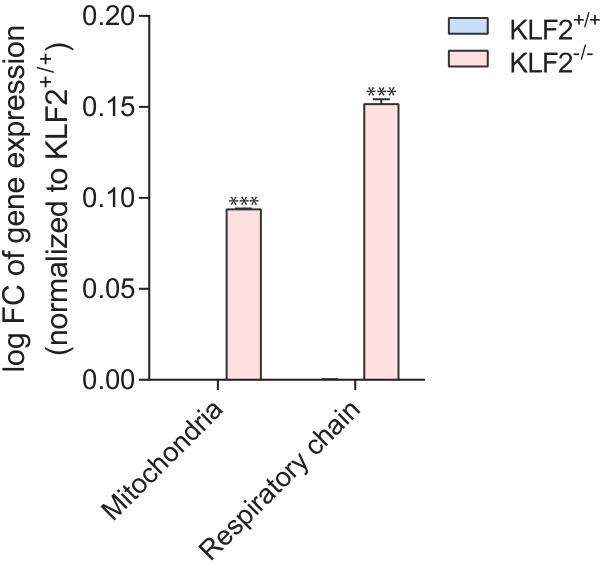
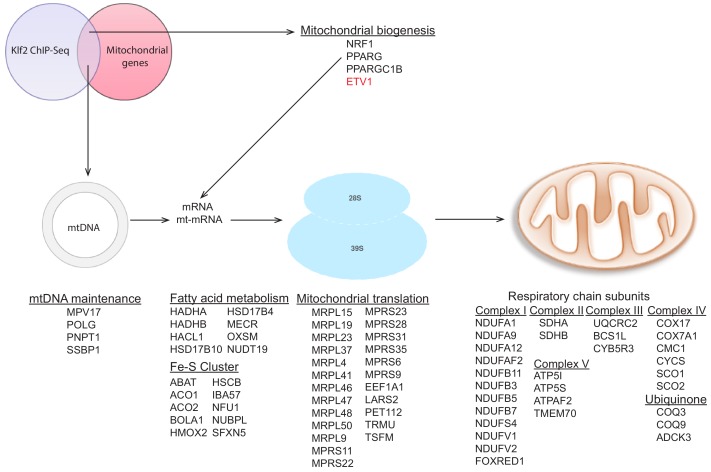
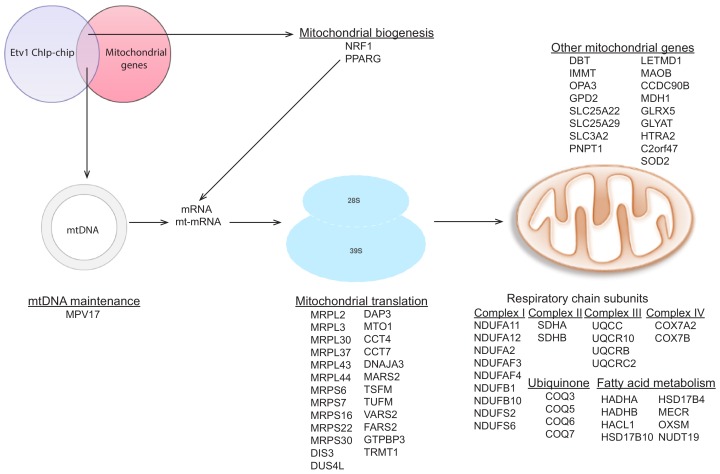
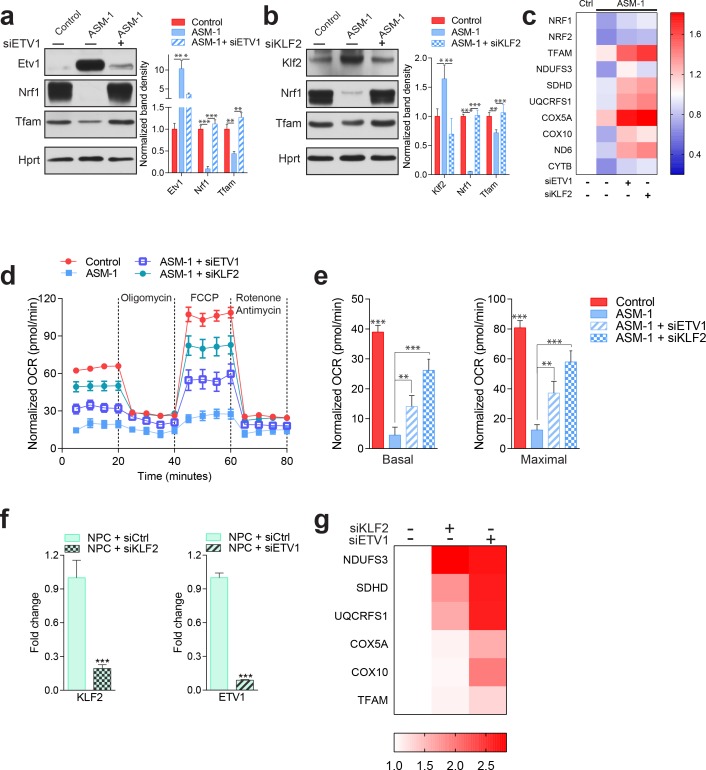
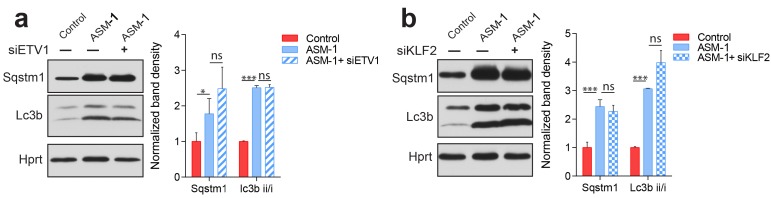
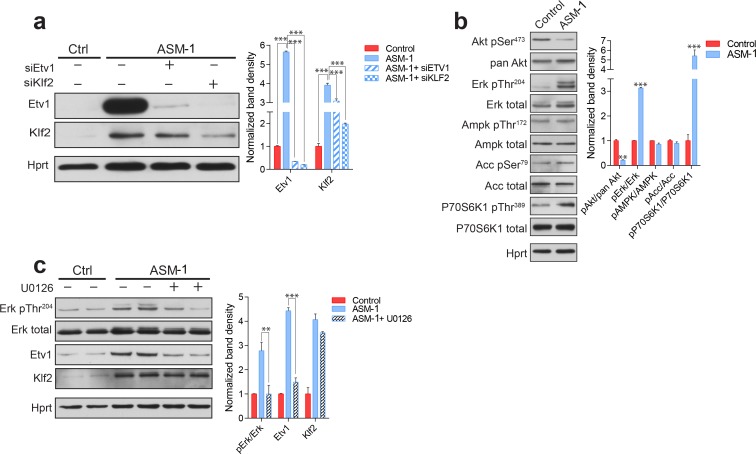
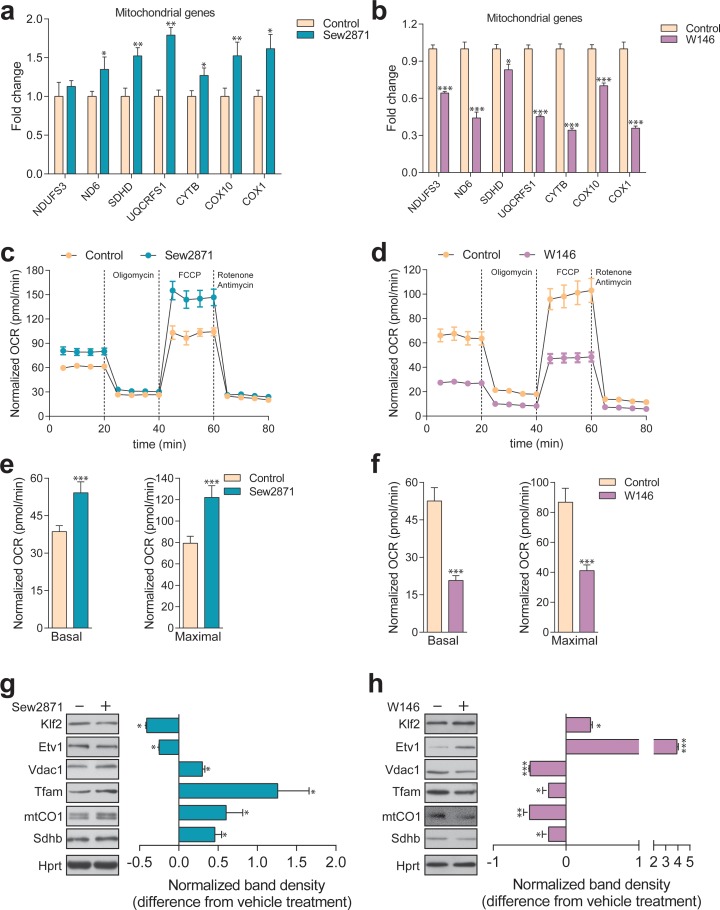
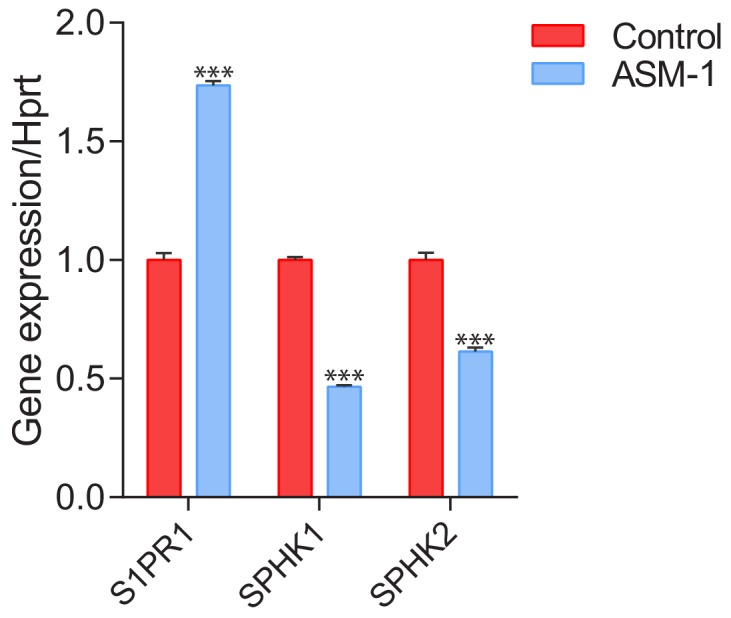
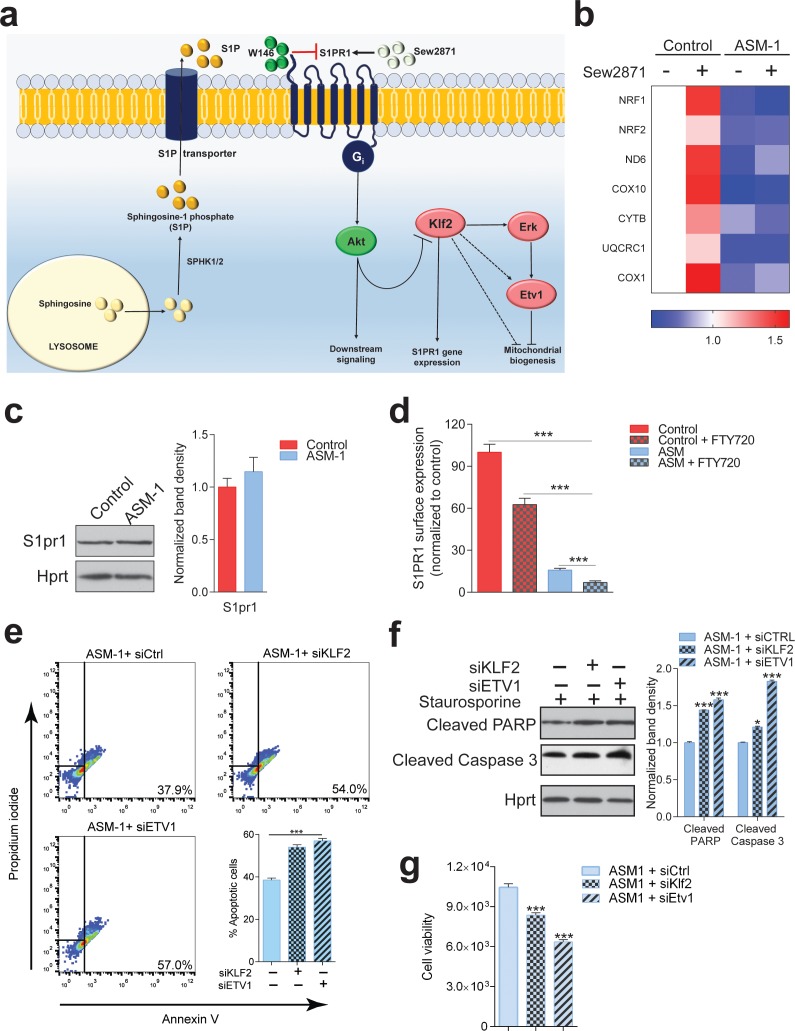
Perturbations in mitochondrial function and homeostasis are pervasive in lysosomal storage diseases, but the underlying mechanisms remain unknown. Here, we report a transcriptional program that represses mitochondrial biogenesis and function in lysosomal storage diseases Niemann-Pick type C (NPC) and acid sphingomyelinase deficiency (ASM), in patient cells and mouse tissues. This mechanism is mediated by the transcription factors KLF2 and ETV1, which are both induced in NPC and ASM patient cells. Mitochondrial biogenesis and function defects in these cells are rescued by the silencing of KLF2 or ETV1. Increased ETV1 expression is regulated by KLF2, while the increase of KLF2 protein levels in NPC and ASM stems from impaired signaling downstream sphingosine-1-phosphate receptor 1 (S1PR1), which normally represses KLF2. In patient cells, S1PR1 is barely detectable at the plasma membrane and thus unable to repress KLF2. This manuscript provides a mechanistic pathway for the prevalent mitochondrial defects in lysosomal storage diseases.
This article has been through an editorial process in which the authors decide how to respond to the issues raised during peer review. The Reviewing Editor's assessment is that all the issues have been addressed (see decision letter).
溶酶体贮积病中普遍存在线粒体功能和动态平衡的紊乱,但潜在机制尚不清楚。在这里,我们报告了一个转录程序,该程序可在溶酶体贮积病尼曼-匹克 C 型(NPC)和酸性鞘磷脂酶缺乏症(ASM)的患者细胞和小鼠组织中抑制线粒体生物发生和功能。该机制由转录因子 KLF2 和 ETV1 介导,这两种转录因子在 NPC 和 ASM 患者细胞中均被诱导。沉默 KLF2 或 ETV1 可挽救这些细胞中线粒体生物发生和功能的缺陷。ETV1 的表达增加受 KLF2 调节,而 NPC 和 ASM 中 KLF2 蛋白水平的增加源于 1-磷酸鞘氨醇受体 1(S1PR1)下游信号转导受损,S1PR1 通常抑制 KLF2。在患者细胞中,S1PR1 几乎检测不到在质膜上,因此无法抑制 KLF2。本文为溶酶体贮积病中普遍存在的线粒体缺陷提供了一种机制途径。
本文经过编辑处理,作者决定如何处理同行评审期间提出的问题。审稿人的评估是所有问题都已得到解决(见评审意见)。