Department of Electrical and Computer Engineering, Drexel University, Philadelphia, Pennsylvania, United States of America.
Department of Computer Science and Engineering, State University of New York at Buffalo, Buffalo, New York, United States of America.
PLoS Comput Biol. 2019 Feb 26;15(2):e1006721. doi: 10.1371/journal.pcbi.1006721. eCollection 2019 Feb.
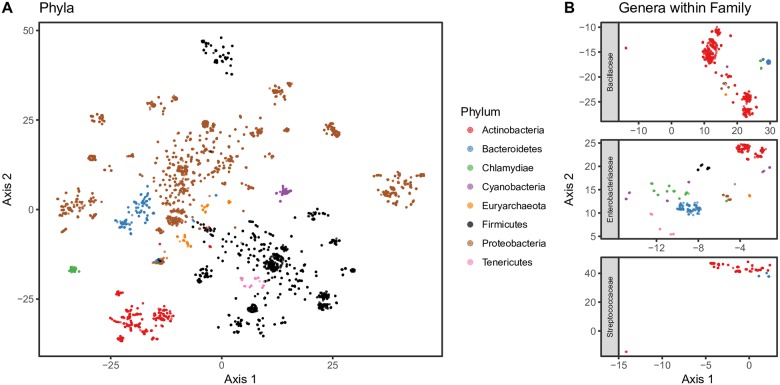
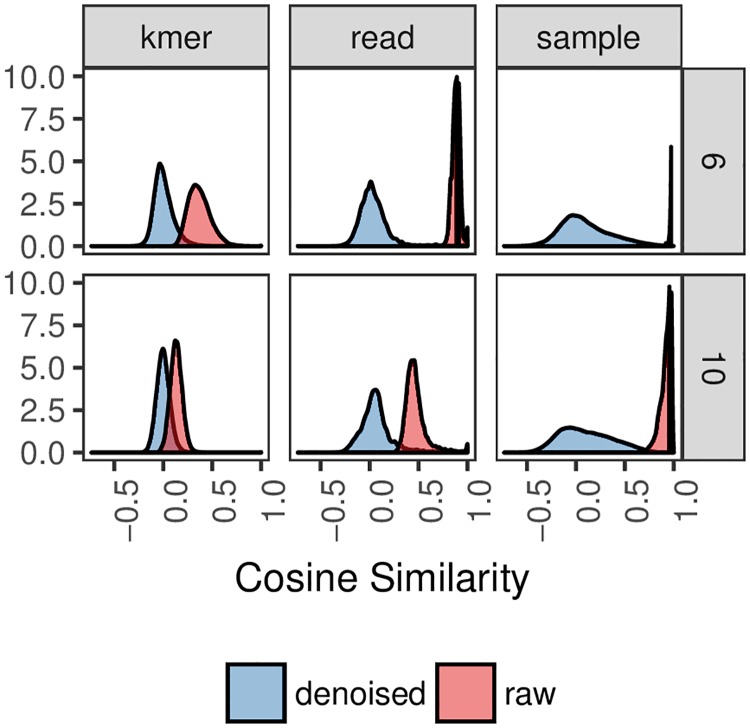
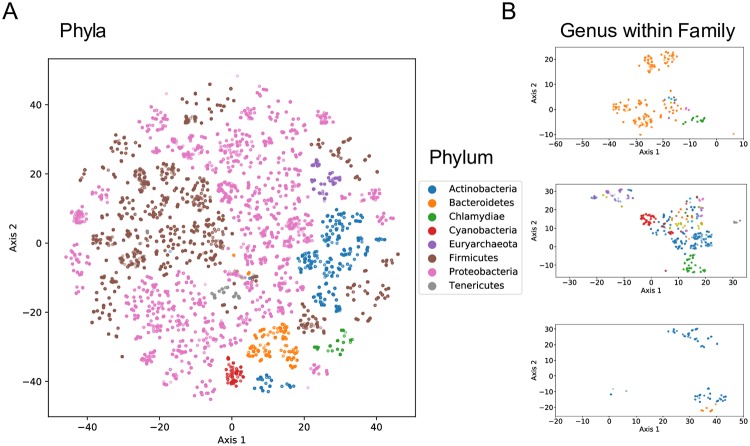
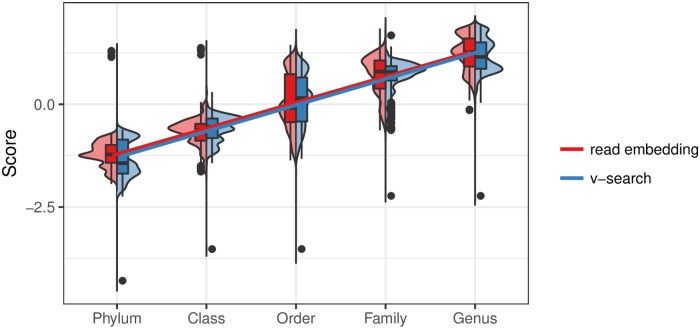
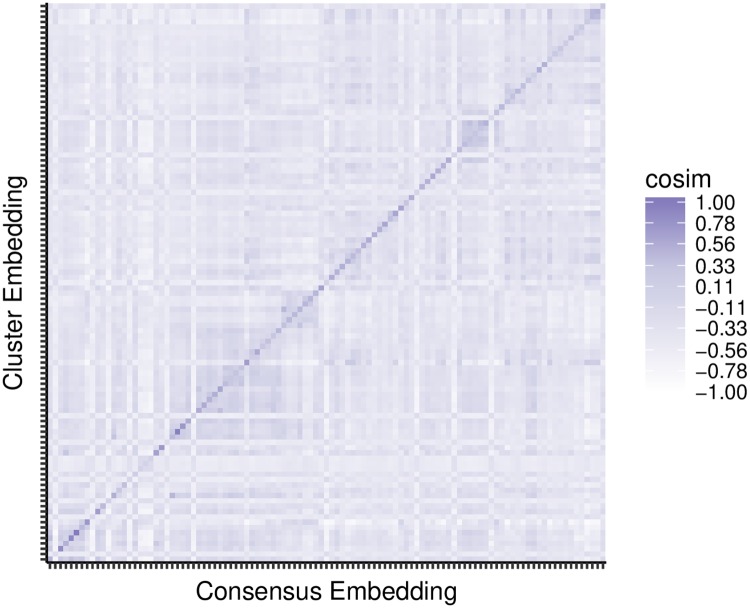
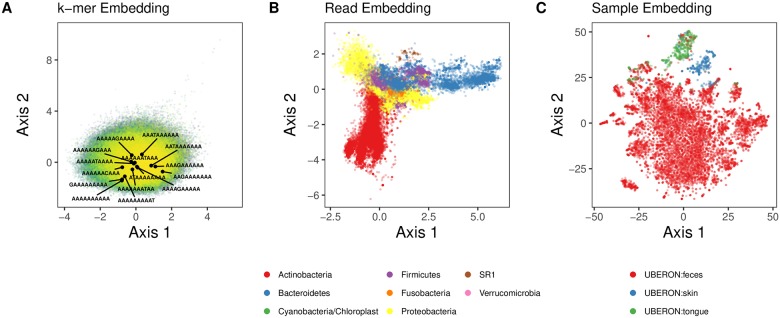
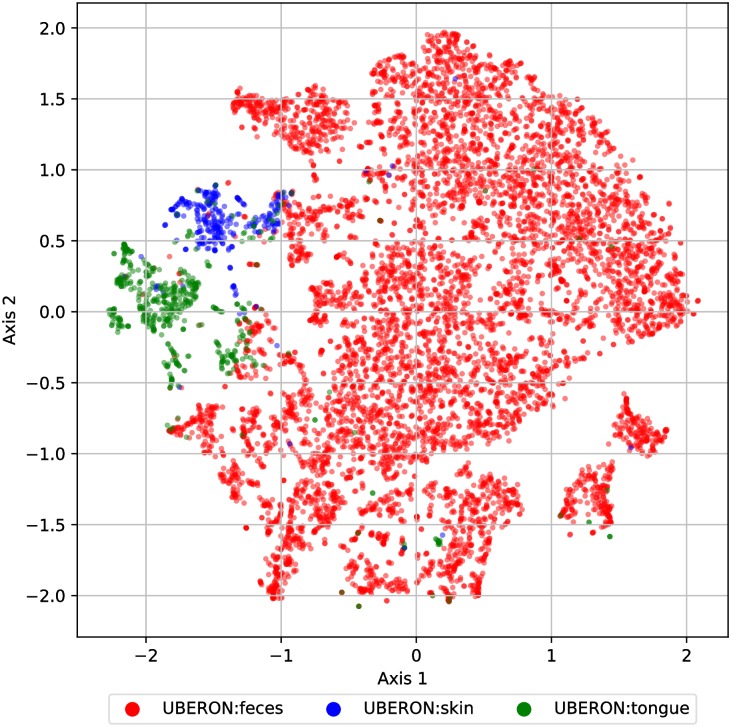
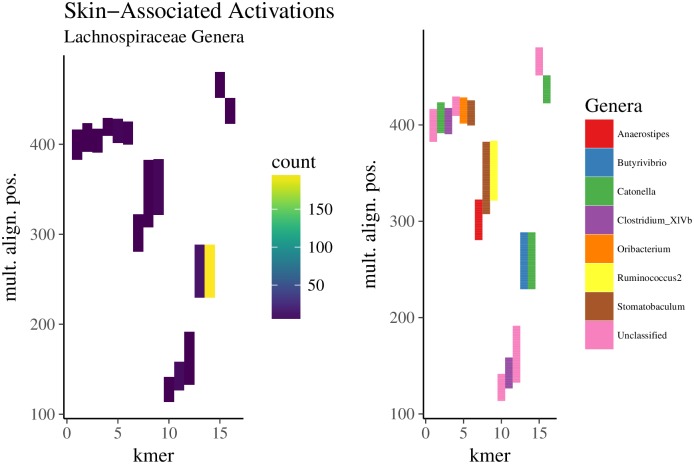
Advances in high-throughput sequencing have increased the availability of microbiome sequencing data that can be exploited to characterize microbiome community structure in situ. We explore using word and sentence embedding approaches for nucleotide sequences since they may be a suitable numerical representation for downstream machine learning applications (especially deep learning). This work involves first encoding ("embedding") each sequence into a dense, low-dimensional, numeric vector space. Here, we use Skip-Gram word2vec to embed k-mers, obtained from 16S rRNA amplicon surveys, and then leverage an existing sentence embedding technique to embed all sequences belonging to specific body sites or samples. We demonstrate that these representations are meaningful, and hence the embedding space can be exploited as a form of feature extraction for exploratory analysis. We show that sequence embeddings preserve relevant information about the sequencing data such as k-mer context, sequence taxonomy, and sample class. Specifically, the sequence embedding space resolved differences among phyla, as well as differences among genera within the same family. Distances between sequence embeddings had similar qualities to distances between alignment identities, and embedding multiple sequences can be thought of as generating a consensus sequence. In addition, embeddings are versatile features that can be used for many downstream tasks, such as taxonomic and sample classification. Using sample embeddings for body site classification resulted in negligible performance loss compared to using OTU abundance data, and clustering embeddings yielded high fidelity species clusters. Lastly, the k-mer embedding space captured distinct k-mer profiles that mapped to specific regions of the 16S rRNA gene and corresponded with particular body sites. Together, our results show that embedding sequences results in meaningful representations that can be used for exploratory analyses or for downstream machine learning applications that require numeric data. Moreover, because the embeddings are trained in an unsupervised manner, unlabeled data can be embedded and used to bolster supervised machine learning tasks.
高通量测序技术的进步增加了微生物组测序数据的可用性,这些数据可用于原位表征微生物组群落结构。我们探索使用单词和句子嵌入方法对核苷酸序列进行编码,因为它们可能是下游机器学习应用(尤其是深度学习)的合适数值表示。这项工作首先将每个序列编码(“嵌入”)为密集的、低维的数字向量空间。在这里,我们使用 Skip-Gram word2vec 嵌入 16S rRNA 扩增子调查获得的 k-mer,然后利用现有的句子嵌入技术来嵌入属于特定身体部位或样本的所有序列。我们证明了这些表示是有意义的,因此可以利用嵌入空间作为探索性分析的特征提取形式。我们表明,序列嵌入保留了有关测序数据的相关信息,例如 k-mer 上下文、序列分类和样本类别。具体来说,序列嵌入空间解决了门、科内属之间的差异。序列嵌入之间的距离与比对身份之间的距离具有相似的性质,并且可以认为嵌入多个序列会生成一个共识序列。此外,嵌入是通用特征,可用于许多下游任务,如分类和样本分类。与使用 OTU 丰度数据相比,使用样本嵌入进行身体部位分类几乎没有性能损失,并且聚类嵌入产生了高保真度的物种聚类。最后,k-mer 嵌入空间捕获了映射到 16S rRNA 基因特定区域并与特定身体部位相对应的独特 k-mer 分布。总之,我们的结果表明,嵌入序列会产生有意义的表示,可以用于探索性分析或需要数值数据的下游机器学习应用。此外,由于嵌入是在无监督的方式下进行训练的,因此可以嵌入未标记的数据并用于增强监督机器学习任务。