Molecular Cell Biomechanics Laboratory, Departments of Bioengineering and Mechanical Engineering, University of California, Berkeley, CA, USA.
Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Lab, Berkeley, CA, USA.
Bioinformatics. 2018 Jul 1;34(13):i32-i42. doi: 10.1093/bioinformatics/bty296.
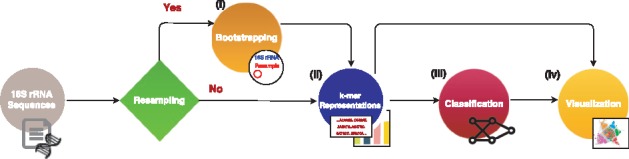
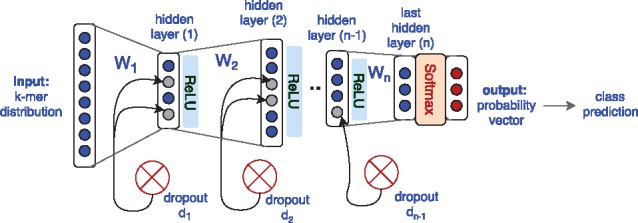
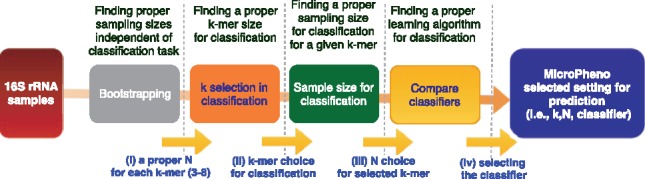
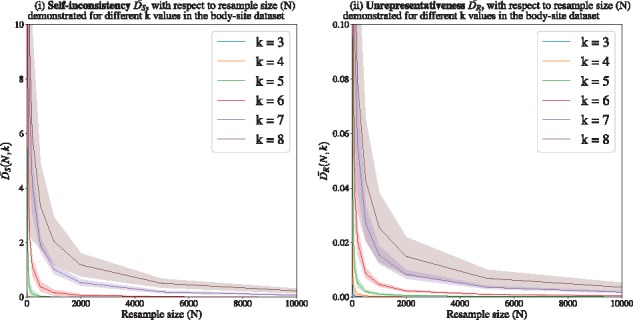
Microbial communities play important roles in the function and maintenance of various biosystems, ranging from the human body to the environment. A major challenge in microbiome research is the classification of microbial communities of different environments or host phenotypes. The most common and cost-effective approach for such studies to date is 16S rRNA gene sequencing. Recent falls in sequencing costs have increased the demand for simple, efficient and accurate methods for rapid detection or diagnosis with proved applications in medicine, agriculture and forensic science. We describe a reference- and alignment-free approach for predicting environments and host phenotypes from 16S rRNA gene sequencing based on k-mer representations that benefits from a bootstrapping framework for investigating the sufficiency of shallow sub-samples. Deep learning methods as well as classical approaches were explored for predicting environments and host phenotypes.
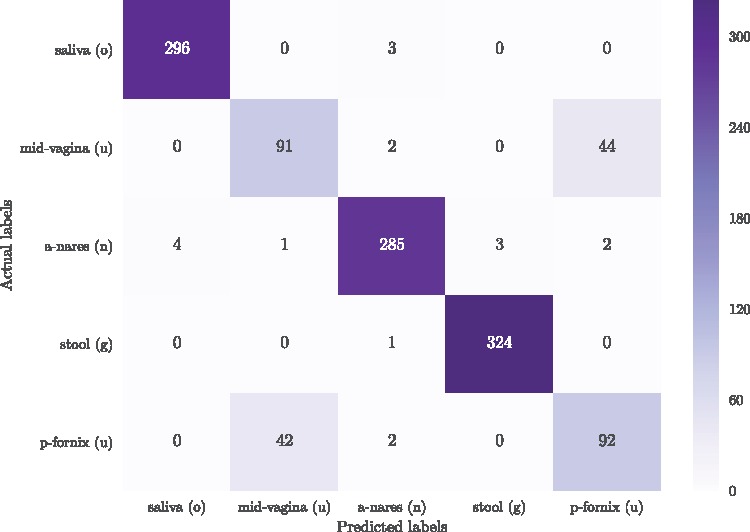
A k-mer distribution of shallow sub-samples outperformed Operational Taxonomic Unit (OTU) features in the tasks of body-site identification and Crohn's disease prediction. Aside from being more accurate, using k-mer features in shallow sub-samples allows (i) skipping computationally costly sequence alignments required in OTU-picking and (ii) provided a proof of concept for the sufficiency of shallow and short-length 16S rRNA sequencing for phenotype prediction. In addition, k-mer features predicted representative 16S rRNA gene sequences of 18 ecological environments, and 5 organismal environments with high macro-F1 scores of 0.88 and 0.87. For large datasets, deep learning outperformed classical methods such as Random Forest and Support Vector Machine.
The software and datasets are available at https://llp.berkeley.edu/micropheno.
Supplementary data are available at Bioinformatics online.
微生物群落在各种生物系统(从人体到环境)的功能和维持中发挥着重要作用。微生物组研究的一个主要挑战是对不同环境或宿主表型的微生物群落进行分类。迄今为止,此类研究最常见和最具成本效益的方法是 16S rRNA 基因测序。最近测序成本的下降增加了对简单、高效和准确方法的需求,这些方法已在医学、农业和法医学中得到证明应用,可用于快速检测或诊断。我们描述了一种基于 k-mer 表示的参考和无比对方法,用于根据 16S rRNA 基因测序预测环境和宿主表型,该方法受益于用于调查浅层子样本充足性的自举框架。我们探索了深度学习方法和经典方法来预测环境和宿主表型。
在体定位识别和克罗恩病预测任务中,浅层子样本的 k-mer 分布优于操作分类单元 (OTU) 特征。除了更准确之外,在浅层子样本中使用 k-mer 特征还可以(i)跳过在 OTU 选择中需要的计算成本高昂的序列比对,(ii)为用于表型预测的短长度 16S rRNA 测序的浅层和短长度的充分性提供了概念验证。此外,k-mer 特征预测了 18 个生态环境和 5 个生物环境的代表性 16S rRNA 基因序列,其宏 F1 分数分别为 0.88 和 0.87。对于大型数据集,深度学习优于随机森林和支持向量机等经典方法。
软件和数据集可在 https://llp.berkeley.edu/micropheno 上获得。
补充数据可在生物信息学在线获得。