Dept. of Informatics, Systems and Communication, University of Milan-Bicocca, 20126, Milan, Italy.
SYSBIO Centre of Systems Biology, 20126, Milan, Italy.
PLoS Comput Biol. 2019 Feb 28;15(2):e1006733. doi: 10.1371/journal.pcbi.1006733. eCollection 2019 Feb.
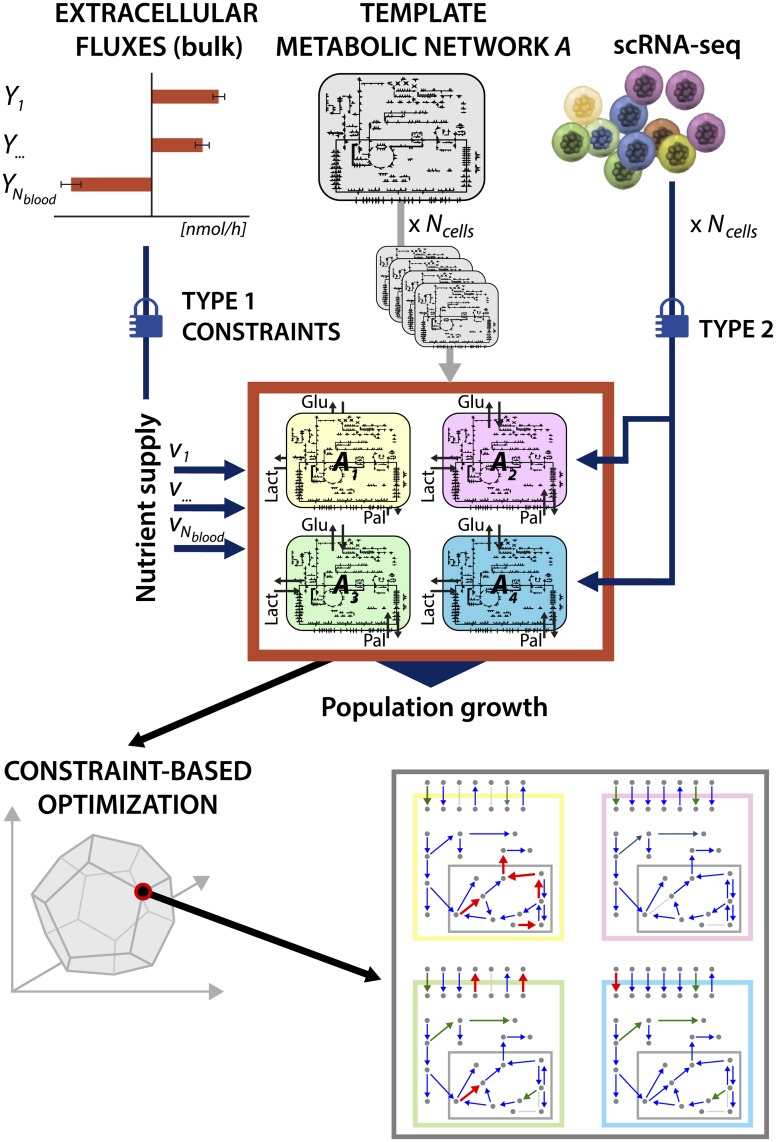
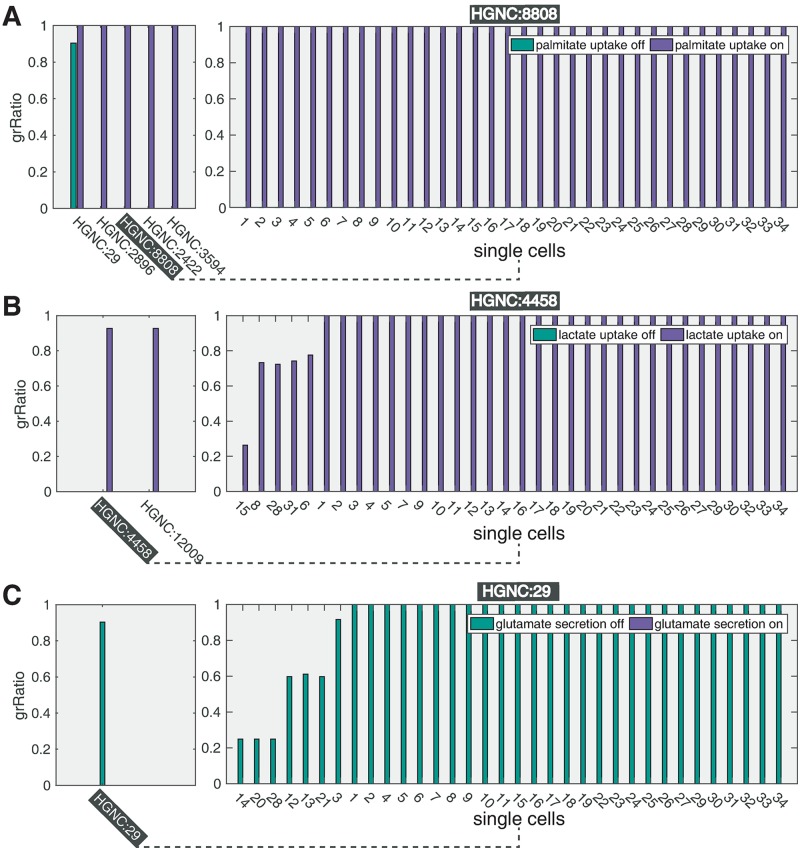
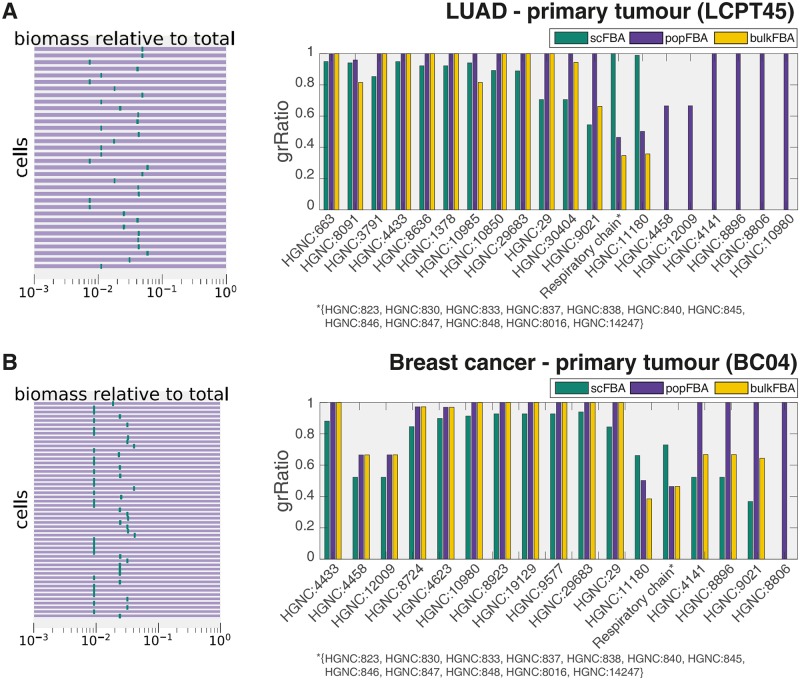
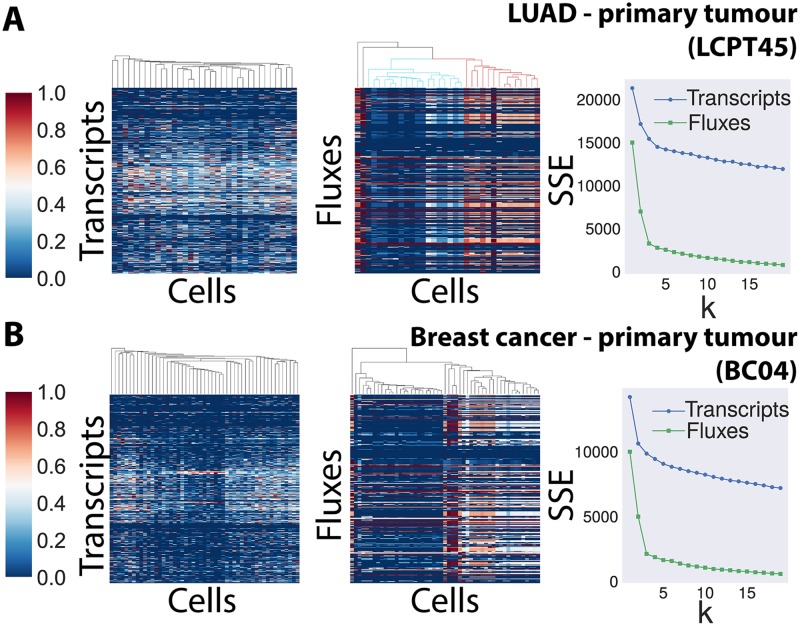
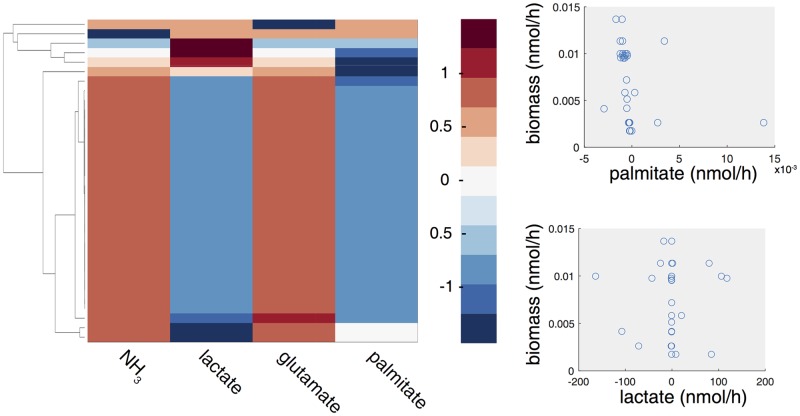
Metabolic reprogramming is a general feature of cancer cells. Regrettably, the comprehensive quantification of metabolites in biological specimens does not promptly translate into knowledge on the utilization of metabolic pathways. By estimating fluxes across metabolic pathways, computational models hold the promise to bridge this gap between data and biological functionality. These models currently portray the average behavior of cell populations however, masking the inherent heterogeneity that is part and parcel of tumorigenesis as much as drug resistance. To remove this limitation, we propose single-cell Flux Balance Analysis (scFBA) as a computational framework to translate single-cell transcriptomes into single-cell fluxomes. We show that the integration of single-cell RNA-seq profiles of cells derived from lung adenocarcinoma and breast cancer patients into a multi-scale stoichiometric model of a cancer cell population: significantly 1) reduces the space of feasible single-cell fluxomes; 2) allows to identify clusters of cells with different growth rates within the population; 3) points out the possible metabolic interactions among cells via exchange of metabolites. The scFBA suite of MATLAB functions is available at https://github.com/BIMIB-DISCo/scFBA, as well as the case study datasets.
代谢重编程是癌细胞的一个普遍特征。遗憾的是,生物样本中代谢物的全面定量分析并不能立即转化为对代谢途径利用的认识。通过估计代谢途径中的通量,计算模型有望弥合数据和生物功能之间的这一差距。这些模型目前描绘了细胞群体的平均行为,但掩盖了肿瘤发生和耐药性所必需的内在异质性。为了消除这一限制,我们提出了单细胞通量平衡分析(scFBA)作为一种计算框架,将单细胞转录组学数据转化为单细胞通量组学数据。我们表明,将源自肺腺癌和乳腺癌患者的细胞的单细胞 RNA-seq 图谱整合到一个癌症细胞群体的多尺度计量模型中:显著地 1)缩小了可行的单细胞通量组学的空间;2)能够在群体内识别出具有不同生长速度的细胞簇;3)通过代谢物交换指出细胞之间可能存在的代谢相互作用。MATLAB 函数的 scFBA 套件可在 https://github.com/BIMIB-DISCo/scFBA 以及案例研究数据集上获得。