Zhou Jian-Guo, Zhong Hua, Zhang Juan, Jin Su-Han, Roudi Raheleh, Ma Hu
Department of Oncology, Affiliated Hospital of Zunyi Medical University, Zunyi, China.
College of Life Sciences, Wuhan University, Wuhan, China.
Front Oncol. 2019 Feb 15;9:78. doi: 10.3389/fonc.2019.00078. eCollection 2019.
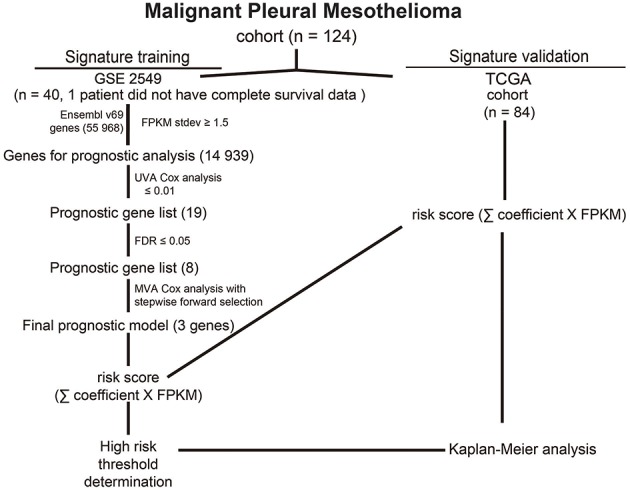
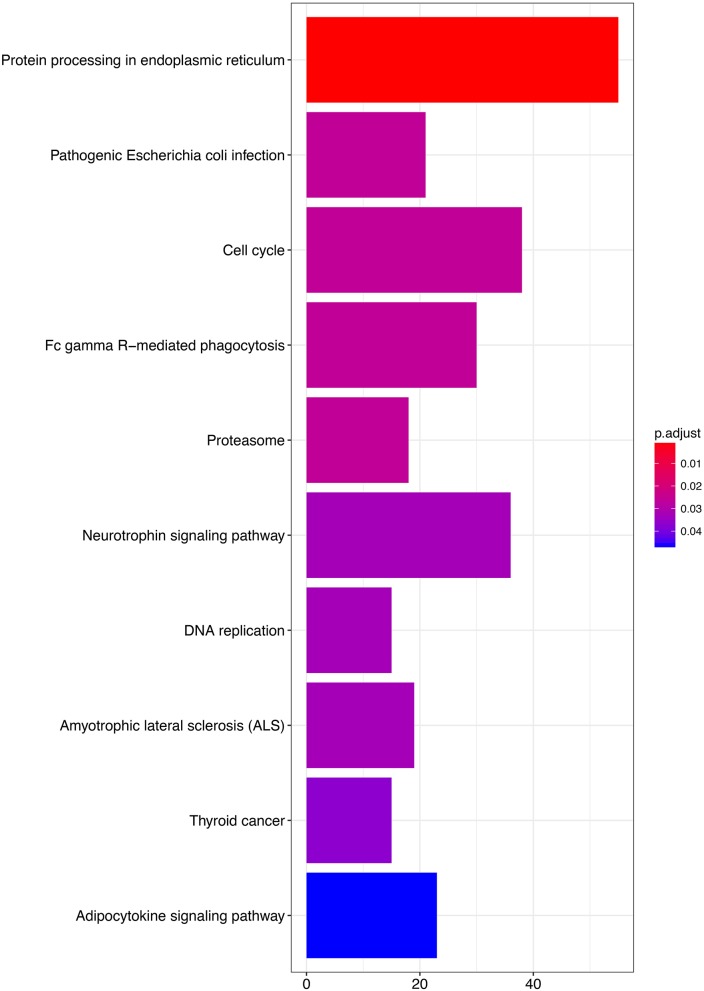
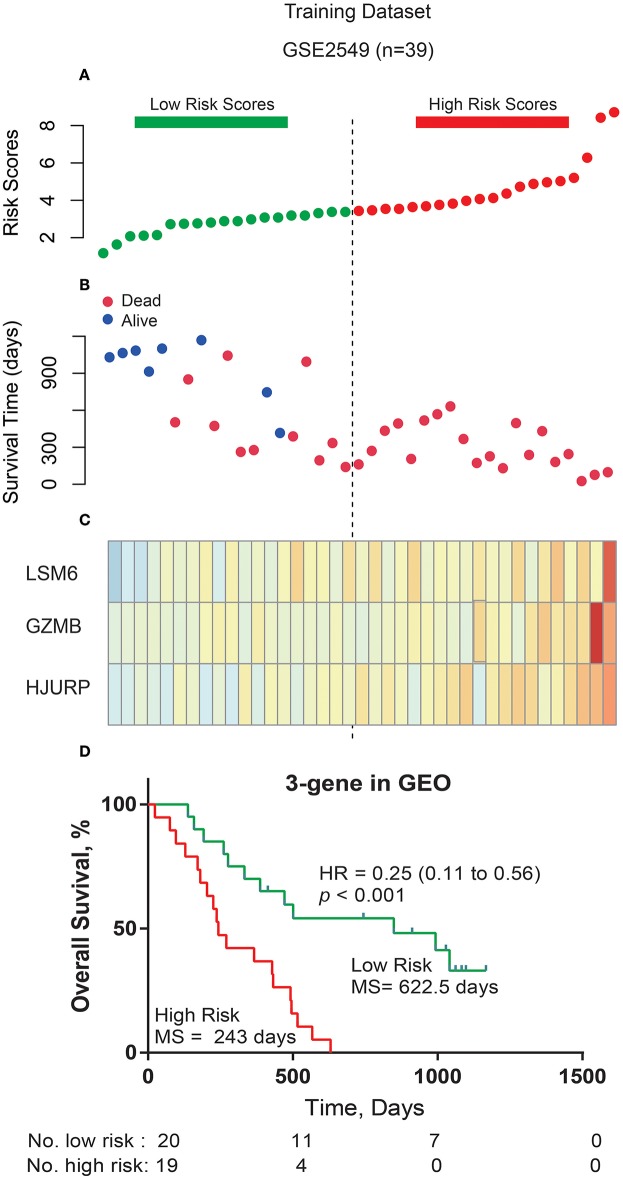
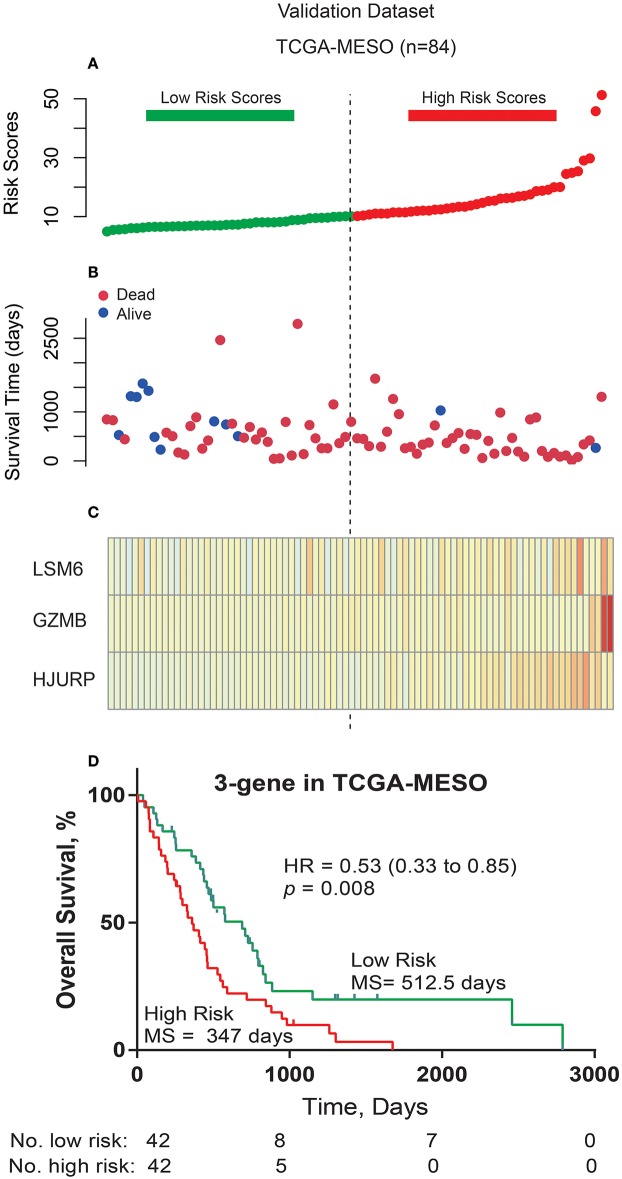
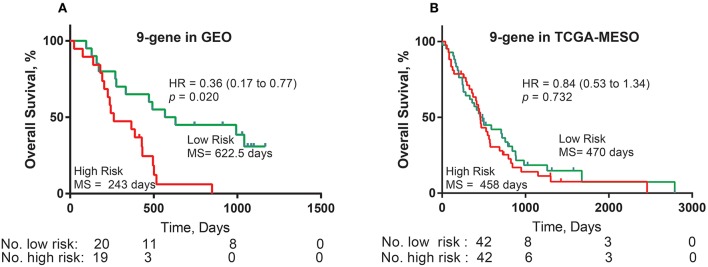
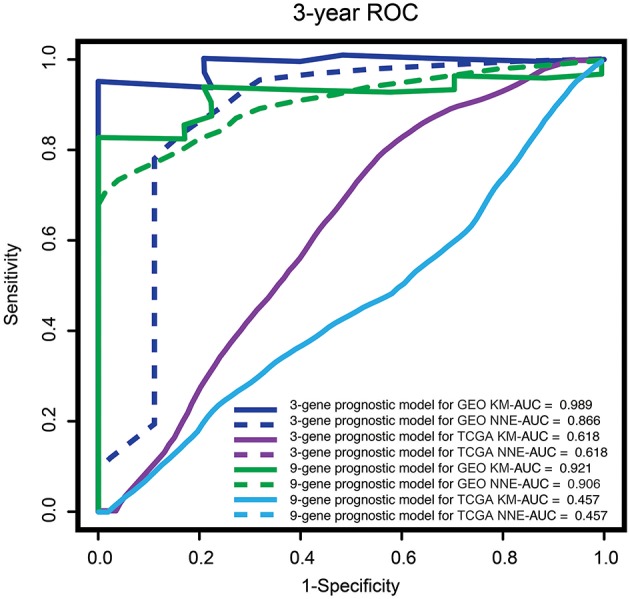
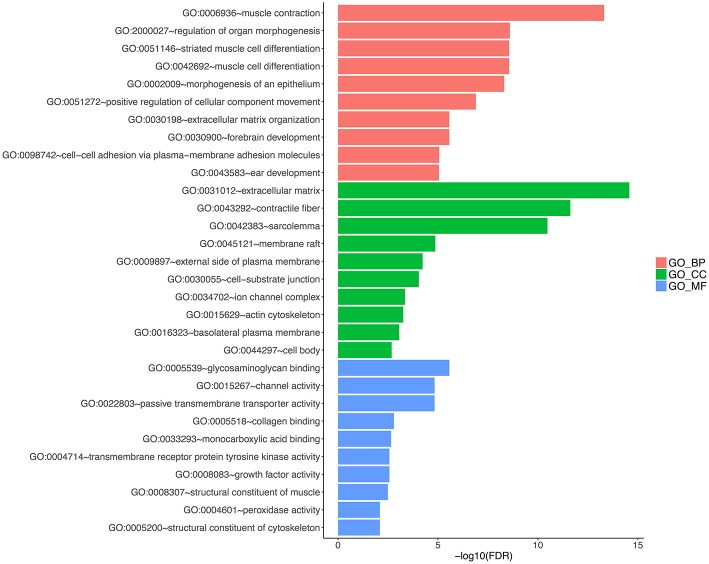
Dysregulated genes play a critical role in the development and progression of cancer, suggesting their potential as novel independent biomarkers for cancer diagnosis and prognosis. Prognostic model-based gene expression profiles are not widely utilized in clinical medicine. We investigated the prognostic significance of an expression profile-based gene signature for outcome prediction in patients with malignant pleural mesothelioma (MPM). The gene expression profiles of a large cohort of patients with MPM were obtained and analyzed by repurposing publicly available microarray data. A gene-based risk score model was developed with the training dataset and then validated with the TCGA-MESO (mesothelioma) dataset. The time-dependent receiver operating characteristic (ROC) curve was used to evaluate the prognostic performance of survival prediction. The biological function of the prognostic genes was predicted using bioinformatics analysis. Three genes in the training dataset (GSE2549) were identified as significantly associated with the overall survival (OS) of patients with MPM and were combined to develop a three-gene prognostic signature to stratify patients into low-risk and high-risk groups. The MPM patients of the training dataset in the low-risk group exhibited longer OS than those in the high-risk group (HR = 0.25, 95% CI = 0.11-0.56, < 0.001). Similar prognostic values for the three-gene signature were observed in the validated TCGA-MESO cohort (HR = 0.53 95% CI = 0.33-0.85, = 0.008). ROC analysis also demonstrated the good performance in predicting 3-year OS in the GEO and TCGA cohorts (KM-AUC for GEO = 0.989, KM-AUC for TCGA = 0.618). The C-statistic for the 3-gene model was 0.761. Validation with TCGA-MESO confirmed the model's ability to discriminate between risk groups in an alternative data set with fair performance (C-statistic: 0.68). Functional enrichment analysis suggested that these three genes may be involved in genetic and epigenetic events with known links to MPM. This study has identified and validated a novel 3-gene model to reliably discriminate patients at high and low risk of death in unselected populations of patients with MPM. Further larger, prospective multi-institutional cohort studies are necessary to validate this model.
失调基因在癌症的发生和发展中起着关键作用,这表明它们有可能成为癌症诊断和预后的新型独立生物标志物。基于预后模型的基因表达谱在临床医学中尚未得到广泛应用。我们研究了基于表达谱的基因特征对恶性胸膜间皮瘤(MPM)患者预后预测的意义。通过重新利用公开可用的微阵列数据,获取并分析了一大群MPM患者的基因表达谱。利用训练数据集开发了基于基因的风险评分模型,然后用TCGA-MESO(间皮瘤)数据集进行验证。采用时间依赖性受试者工作特征(ROC)曲线来评估生存预测的预后性能。使用生物信息学分析预测预后基因的生物学功能。在训练数据集(GSE2549)中,有三个基因被确定与MPM患者的总生存期(OS)显著相关,并将它们组合起来开发了一个三基因预后特征,以将患者分为低风险和高风险组。训练数据集中低风险组的MPM患者的OS比高风险组的患者更长(HR = 0.25,95% CI = 0.11 - 0.56,< 0.001)。在经过验证的TCGA-MESO队列中观察到了三基因特征类似的预后价值(HR = 0.53,95% CI = 0.33 - 0.85, = 0.008)。ROC分析还表明,在GEO和TCGA队列中,该特征在预测3年OS方面表现良好(GEO的KM-AUC = 0.989,TCGA的KM-AUC = 0.618)。三基因模型的C统计量为0.761。用TCGA-MESO进行验证证实了该模型在另一个数据集中区分风险组的能力,性能尚可(C统计量:0.68)。功能富集分析表明,这三个基因可能参与了与MPM有已知联系的遗传和表观遗传事件。本研究确定并验证了一种新型三基因模型,该模型能够可靠地区分未选择的MPM患者群体中高死亡风险和低死亡风险的患者。需要进一步开展更大规模的前瞻性多机构队列研究来验证该模型。