Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, CA, United States of America.
Department of Biochemistry and Biophysics, University of California, San Francisco, San Francisco, CA, United States of America.
PLoS One. 2019 Mar 13;14(3):e0212735. doi: 10.1371/journal.pone.0212735. eCollection 2019.
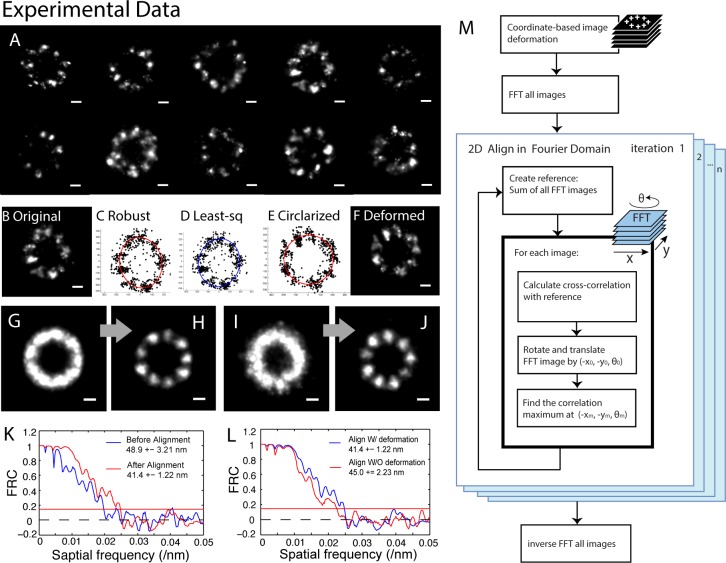
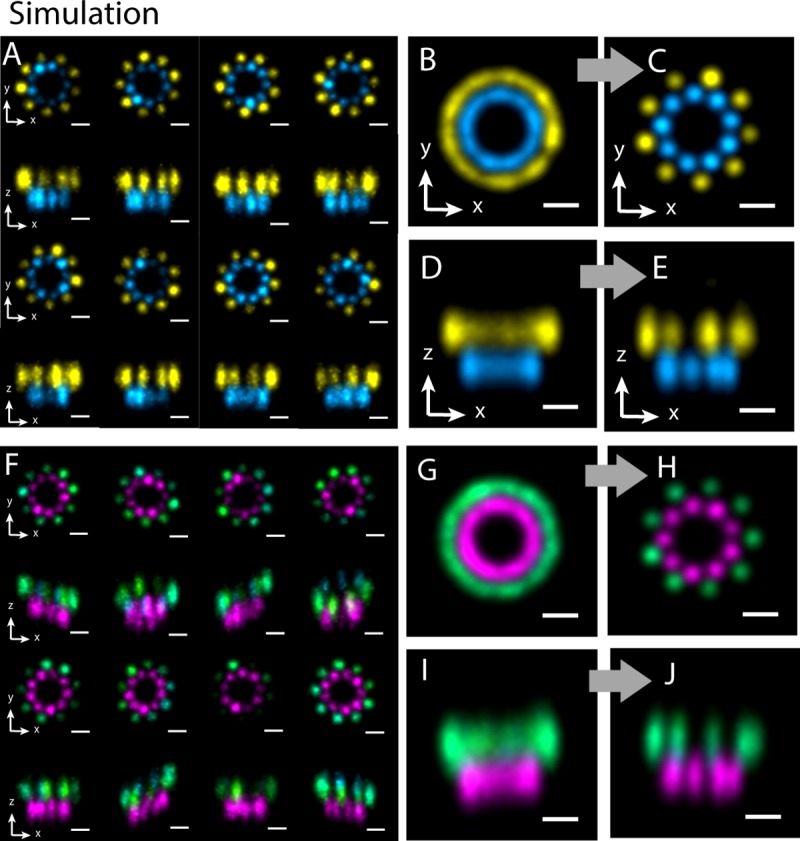
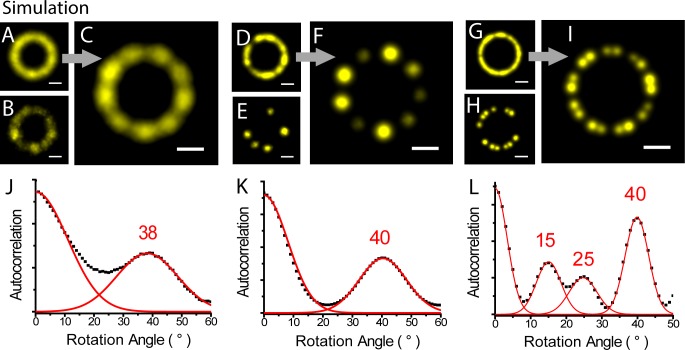
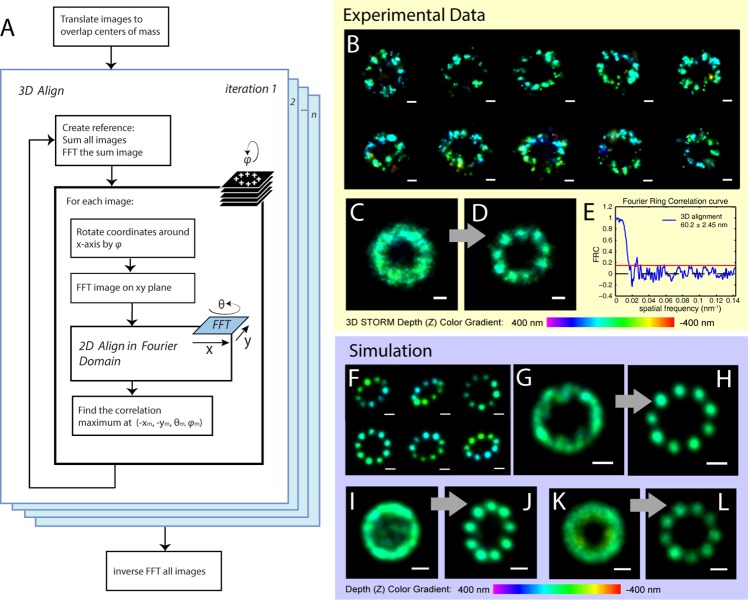
Due to low labeling efficiency and structural heterogeneity in fluorescence-based single-molecule localization microscopy (SMLM), image alignment and quantitative analysis is often required to make accurate conclusions on the spatial relationships between proteins. Cryo-electron microscopy (EM) image alignment procedures have been applied to average structures taken with super-resolution microscopy. However, unlike cryo-EM, the much larger cellular structures analyzed by super-resolution microscopy are often heterogeneous, resulting in misalignment. And the light-microscopy image library is much smaller, which makes classification challenging. To overcome these two challenges, we developed a method to deform semi-flexible ring-shaped structures and then align the 3D structures without classification. These algorithms can register semi-flexible structures with an accuracy of several nanometers in short computation time and with greatly reduced memory requirements. We demonstrated our methods by aligning experimental Stochastic Optical Reconstruction Microscopy (STORM) images of ciliary distal appendages and simulated structures. Symmetries, dimensions, and locations of protein complexes in 3D are revealed by the alignment and averaging for heterogeneous, tilted, and under-labeled structures.
由于基于荧光的单分子定位显微镜(SMLM)的标记效率低和结构异质性,通常需要进行图像对齐和定量分析,才能对蛋白质之间的空间关系做出准确的结论。已经将冷冻电子显微镜(cryo-EM)图像对齐程序应用于超分辨率显微镜获得的平均结构。然而,与 cryo-EM 不同,超分辨率显微镜分析的细胞结构通常较大且具有异质性,导致对齐不准确。并且,用于分类的光显微镜图像库要小得多。为了克服这两个挑战,我们开发了一种方法来变形半柔性环形结构,然后在不分类的情况下对齐 3D 结构。这些算法可以在短的计算时间内以几个纳米的精度注册半柔性结构,并且大大减少了内存需求。我们通过对齐实验性的纤毛远端附属物的随机光学重建显微镜(STORM)图像和模拟结构来证明我们的方法。通过对齐和平均处理具有异质性、倾斜和标记不足的结构,可以揭示蛋白质复合物在 3D 中的对称性、尺寸和位置。