Department of Biostatistics, School of Public Health, Nanjing Medical University, 101 Longmian Avenue, Nanjing, Jiangsu, People's Republic of China.
Department of Pharmacy Informatics, School of Science, China Pharmaceutical University, 24 Tongjia Xiang, Nanjing , Jiangsu, People's Republic of China.
BMC Genet. 2019 Mar 19;20(1):36. doi: 10.1186/s12863-019-0739-7.
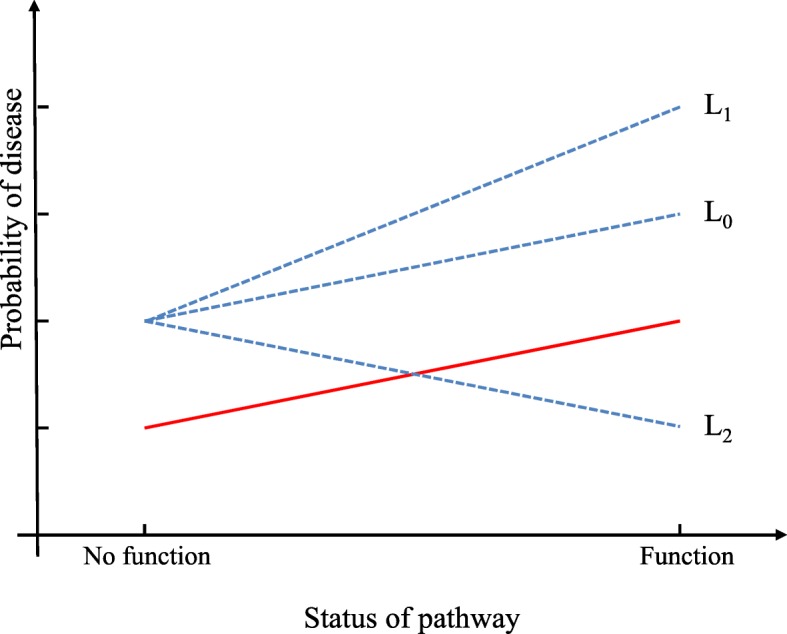
RNA sequencing (RNA-seq) technology has identified multiple differentially expressed (DE) genes associated to complex disease, however, these genes only explain a modest part of variance. Omnigenic model assumes that disease may be driven by genes with indirect relevance to disease and be propagated by functional pathways. Here, we focus on identifying the interactions between the external genes and functional pathways, referring to gene-pathway interactions (GPIs). Specifically, relying on the relationship between the garrote kernel machine (GKM) and variance component test and permutations for the empirical distributions of score statistics, we propose an efficient analysis procedure as Permutation based gEne-pAthway interaction identification in binary phenotype (PEA).
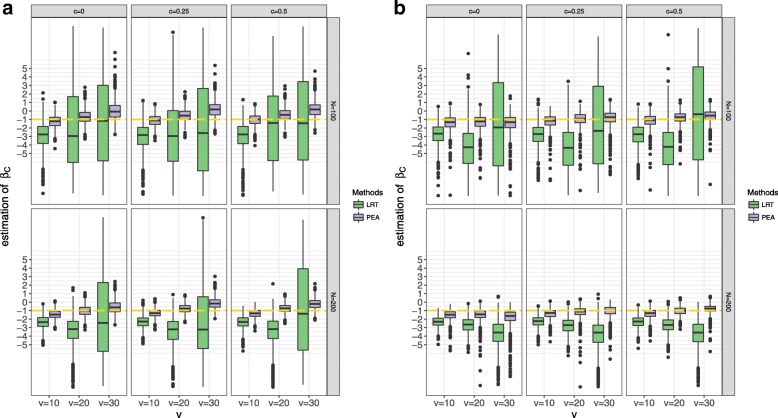
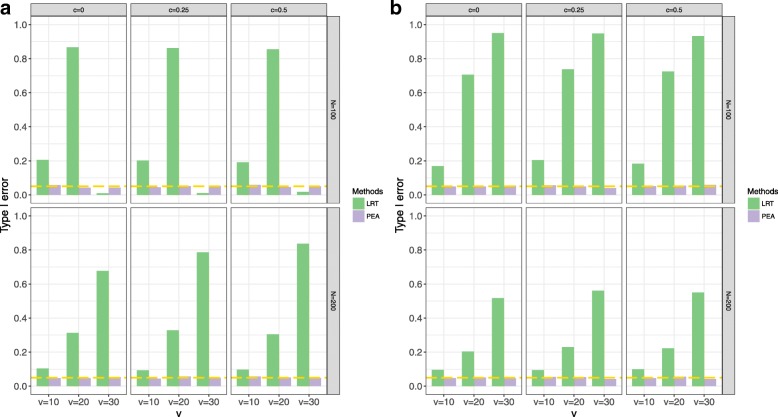
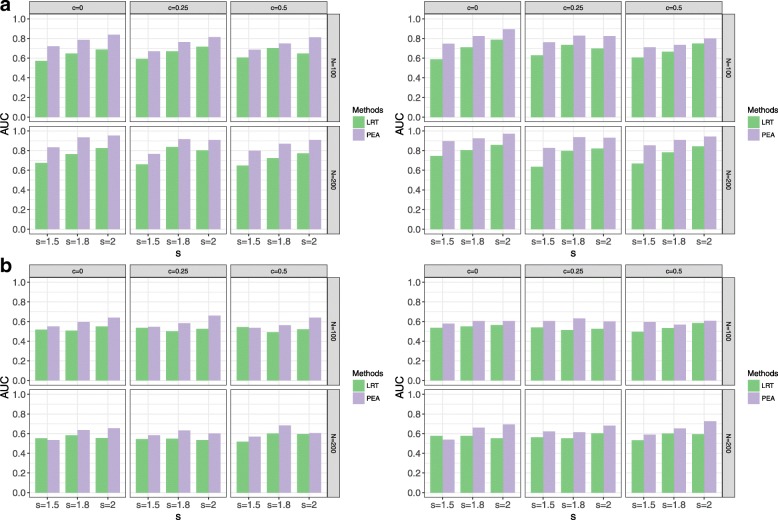
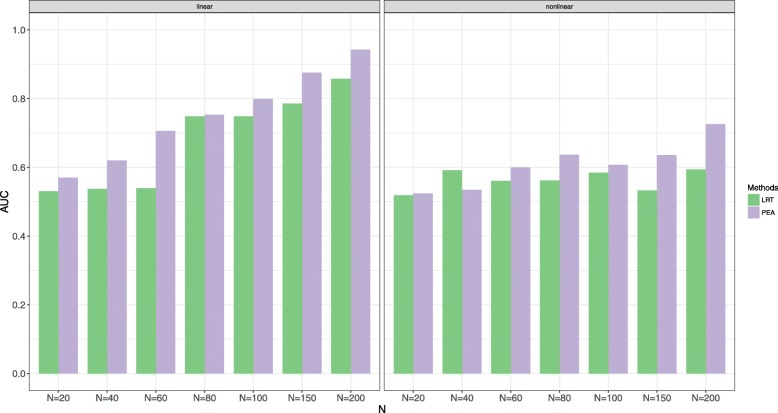
Various simulations show that PEA has well-calibrated type I error rates and higher power than the traditional likelihood ratio test (LRT). In addition, we perform the gene set enrichment algorithms and PEA to identifying the GPIs from a pan-cancer data (GES68086). These GPIs and genes possibly further illustrate the potential etiology of cancers, most of which are identified and some external genes and significant pathways are consistent with previous studies.
PEA is an efficient tool for identifying the GPIs from RNA-seq data. It can be further extended to identify the interactions between one variable and one functional set of other omics data for binary phenotypes.
RNA 测序(RNA-seq)技术已经确定了与复杂疾病相关的多个差异表达(DE)基因,但这些基因仅能解释部分方差。全基因组模型假设疾病可能由与疾病间接相关的基因驱动,并通过功能途径传播。在这里,我们专注于识别外部基因与功能途径之间的相互作用,即基因-途径相互作用(GPIs)。具体来说,我们依赖于绞索核机器(GKM)和方差分量检验之间的关系,以及分数统计量经验分布的置换,提出了一种有效的分析程序,即基于置换的二元表型基因-途径相互作用识别(PEA)。
各种模拟表明,PEA 具有良好校准的Ⅰ型错误率和比传统似然比检验(LRT)更高的功效。此外,我们对泛癌数据(GES68086)进行了基因集富集算法和 PEA 以识别 GPIs。这些 GPIs 和基因可能进一步说明了癌症的潜在病因,其中大部分是已识别的,一些外部基因和重要途径与之前的研究一致。
PEA 是一种从 RNA-seq 数据中识别 GPIs 的有效工具。它可以进一步扩展,以识别二元表型中一个变量与其他组学数据中一组功能的相互作用。