He Zhiqiang, Pan Yueyun, Shao Fang, Wang Hui
Department of Biostatistics, School of Public Health, Nanjing Medical University, Nanjing, China.
First Clinical Medical College, Nanjing Medical University, Nanjing, China.
Front Genet. 2021 Feb 5;12:616686. doi: 10.3389/fgene.2021.616686. eCollection 2021.
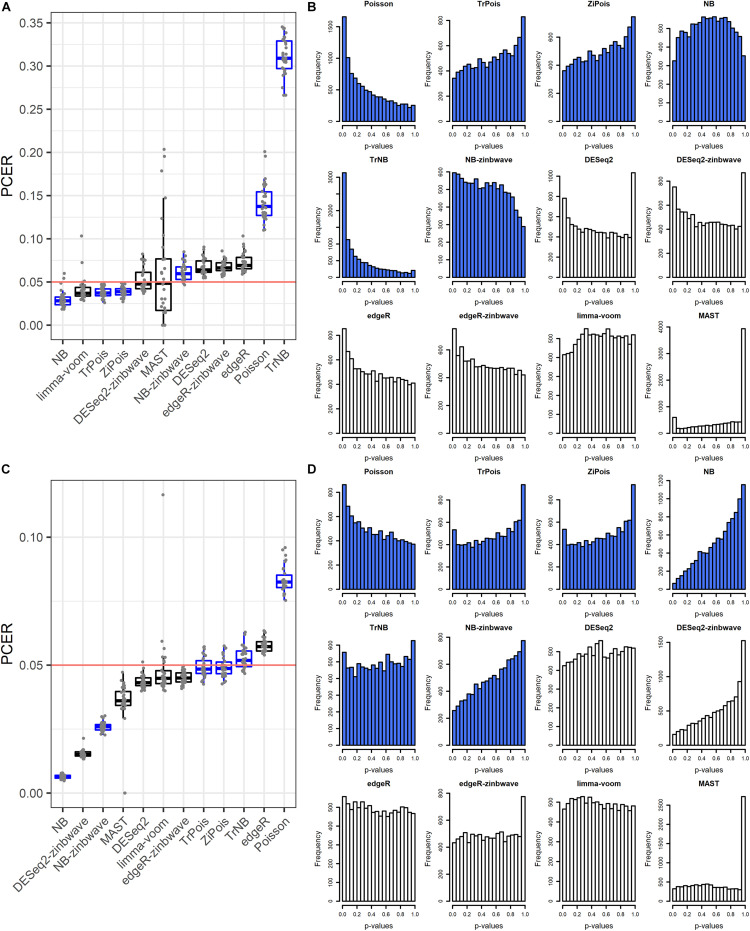
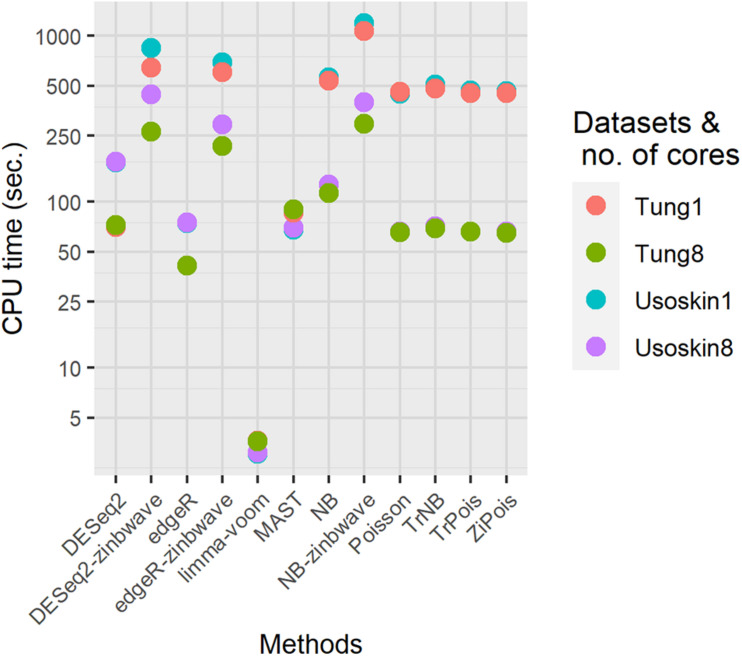
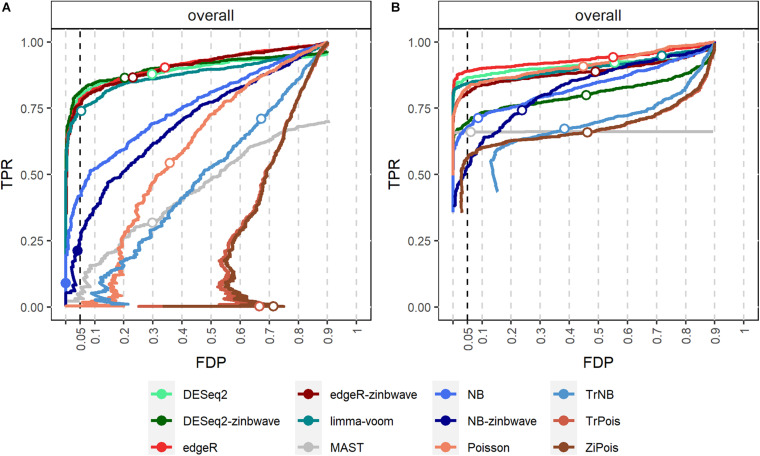
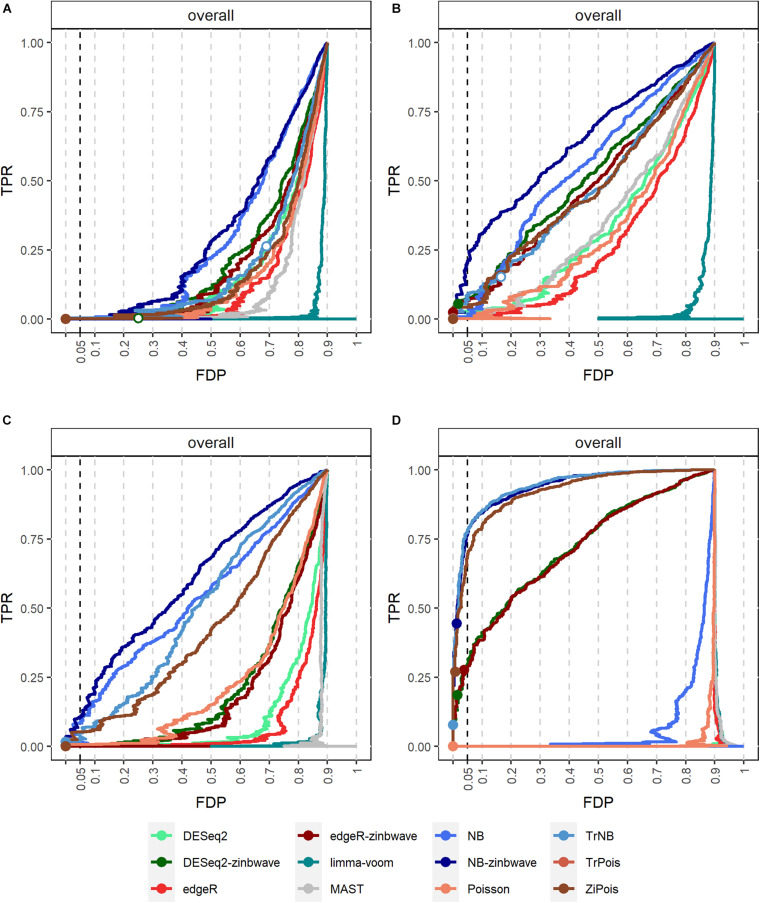
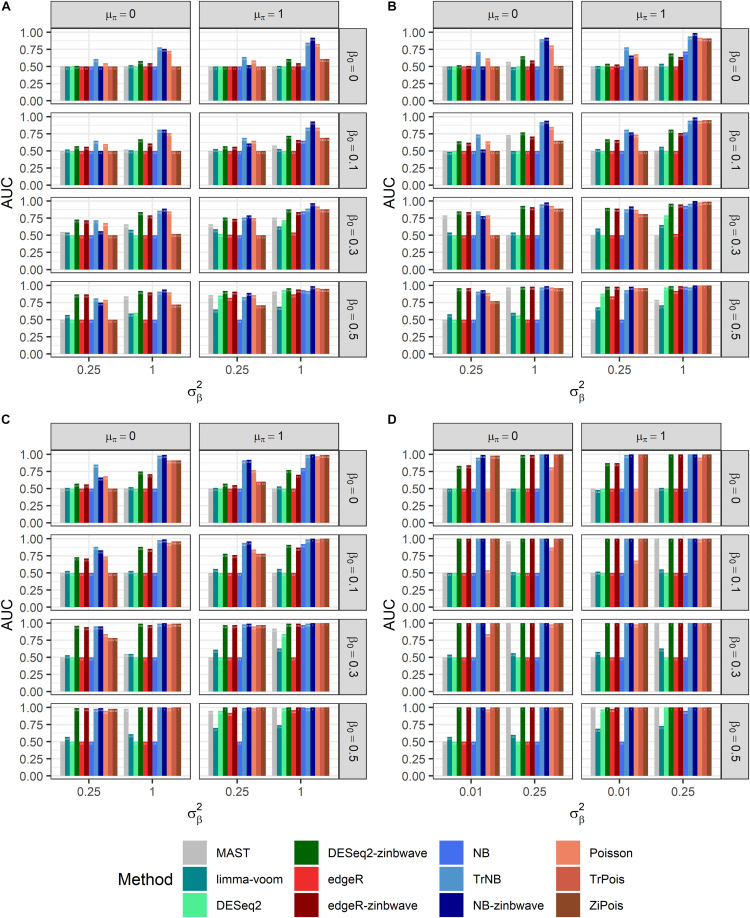
Single cell RNA sequencing (scRNA-seq) allows quantitative measurement and comparison of gene expression at the resolution of single cells. Ignoring the batch effects and zero inflation of scRNA-seq data, many proposed differentially expressed (DE) methods might generate bias. We propose a method, single cell mixed model score tests (scMMSTs), to efficiently identify DE genes of scRNA-seq data with batch effects using the generalized linear mixed model (GLMM). scMMSTs treat the batch effect as a random effect. For zero inflation, scMMSTs use a weighting strategy to calculate observational weights for counts independently under zero-inflated and zero-truncated distributions. Counts data with calculated weights were subsequently analyzed using weighted GLMMs. The theoretical null distributions of the score statistics were constructed by mixed Chi-square distributions. Intensive simulations and two real datasets were used to compare edgeR-zinbwave, DESeq2-zinbwave, and scMMSTs. Our study demonstrates that scMMSTs, as supplement to standard methods, are advantageous to define DE genes of zero-inflated scRNA-seq data with batch effects.
单细胞RNA测序(scRNA-seq)能够在单细胞分辨率下对基因表达进行定量测量和比较。忽略scRNA-seq数据的批次效应和零膨胀问题,许多已提出的差异表达(DE)方法可能会产生偏差。我们提出了一种方法,即单细胞混合模型得分检验(scMMSTs),以使用广义线性混合模型(GLMM)有效地识别具有批次效应的scRNA-seq数据中的DE基因。scMMSTs将批次效应视为随机效应。对于零膨胀问题,scMMSTs使用加权策略在零膨胀和零截断分布下独立计算计数的观测权重。随后使用加权GLMM对具有计算权重的计数数据进行分析。得分统计量的理论零分布由混合卡方分布构建。通过密集模拟和两个真实数据集对edgeR-zinbwave、DESeq2-zinbwave和scMMSTs进行了比较。我们的研究表明,作为标准方法的补充,scMMSTs有利于定义具有批次效应的零膨胀scRNA-seq数据中的DE基因。