Division of Rheumatology and Immunology, Department of Medicine, Medical University of South Carolina, Charleston, SC, USA.
Department of Public Health Sciences, Medical University of South Carolina, Charleston, SC, USA.
Clin Epigenetics. 2019 Apr 4;11(1):58. doi: 10.1186/s13148-019-0652-y.
Systemic sclerosis (SSc) is a rare autoimmune fibrosing disease with an incompletely understood genetic and non-genetic etiology. Defining its etiology is important to allow the development of effective predictive, preventative, and therapeutic strategies. We conducted this epigenomic study to investigate the contributions of DNA methylation to the etiology of SSc while minimizing confounding due to genetic heterogeneity.
Genomic methylation in whole blood from 27 twin pairs discordant for SSc was assayed over 450 K CpG sites. In silico integration with reported differentially methylated cytosines, differentially expressed genes, and regulatory annotation was conducted to validate and interpret the results.
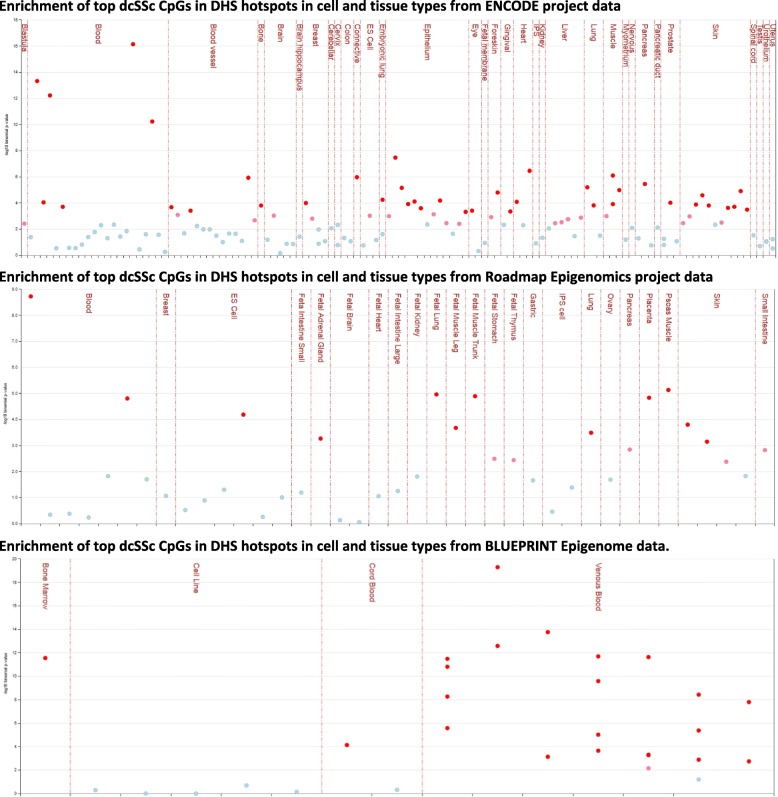
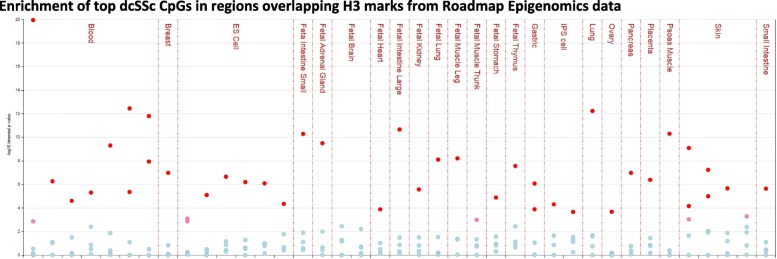
A total of 153 unique cytosines in limited cutaneous SSc (lcSSc) and 266 distinct sites in diffuse cutaneous SSc (dcSSc) showed suggestive differential methylation levels in affected twins. Integration with available data revealed 76 CpGs that were also differentially methylated in blood cells from lupus patients, suggesting their role as potential epigenetic blood biomarkers of autoimmunity. It also revealed 27 genes with concomitant differential expression in blood from SSc patients, including IFI44L and RSAD2. Regulatory annotation revealed that dcSSc-associated CpGs (but not lcSSc) are enriched at Encyclopedia of DNA Elements-, Roadmap-, and BLUEPRINT-derived regulatory regions, supporting their potential role in disease presentation. Notably, the predominant enrichment of regulatory regions in monocytes and macrophages is consistent with the role of these cells in fibrosis, suggesting that the observed cellular dysregulation might be, at least partly, due to altered epigenetic mechanisms of these cells in dcSSc.
These data implicate epigenetic changes in the pathogenesis of SSc and suggest functional mechanisms in SSc etiology.
系统性硬化症(SSc)是一种罕见的自身免疫性纤维性疾病,其遗传和非遗传病因尚未完全阐明。明确其病因对于开发有效的预测、预防和治疗策略非常重要。我们进行了这项表观基因组研究,旨在调查 DNA 甲基化对 SSc 病因的贡献,同时最大限度地减少遗传异质性引起的混杂。
对 27 对系统性硬化症双胞胎中不符合系统性硬化症的 27 对双胞胎的全血进行了 450K 个 CpG 位点的基因组甲基化检测。通过与报道的差异甲基化胞嘧啶、差异表达基因和调控注释进行计算整合,对结果进行验证和解释。
局限性皮肤型 SSc(lcSSc)中共有 153 个独特的胞嘧啶,弥漫性皮肤型 SSc(dcSSc)中共有 266 个独特的位点,在受影响的双胞胎中显示出提示性的差异甲基化水平。与现有数据的整合表明,76 个 CpG 在狼疮患者的血细胞中也表现出差异甲基化,这表明它们可能作为自身免疫性的潜在表观遗传血液生物标志物。它还揭示了 27 个在 SSc 患者血液中同时存在差异表达的基因,包括 IFI44L 和 RSAD2。调控注释表明,dcSSc 相关的 CpG(而不是 lcSSc)在 DNA 元素百科全书、路线图和蓝图衍生的调控区域中富集,支持它们在疾病表现中的潜在作用。值得注意的是,单核细胞和巨噬细胞中主要的调控区域富集与这些细胞在纤维化中的作用一致,这表明观察到的细胞失调至少部分是由于这些细胞在 dcSSc 中的表观遗传机制改变所致。
这些数据表明 DNA 甲基化变化在 SSc 的发病机制中起作用,并提出了 SSc 病因学中的功能机制。