Wang Yanbo, Zhang Weixi, Ding Changjun, Zhang Bingyu, Huang Qinjun, Huang Rongfeng, Su Xiaohua
State Key Laboratory of Tree Genetics and Breeding, Research Institute of Forestry, Chinese Academy of Forestry, Beijing, China.
Key Laboratory of Tree Breeding and Cultivation, National Forestry and Grassland Administration, Beijing, China.
Front Microbiol. 2019 Mar 26;10:588. doi: 10.3389/fmicb.2019.00588. eCollection 2019.
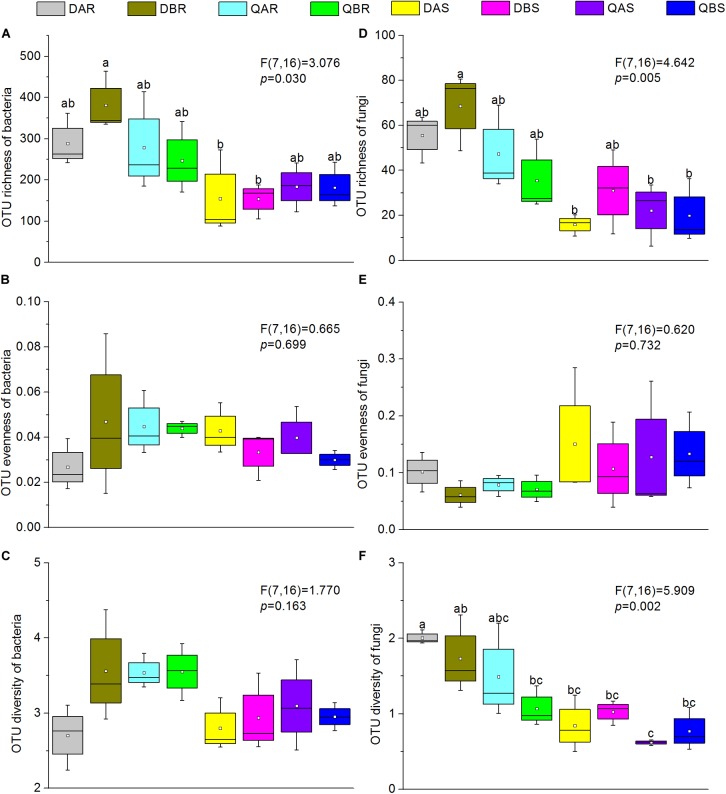
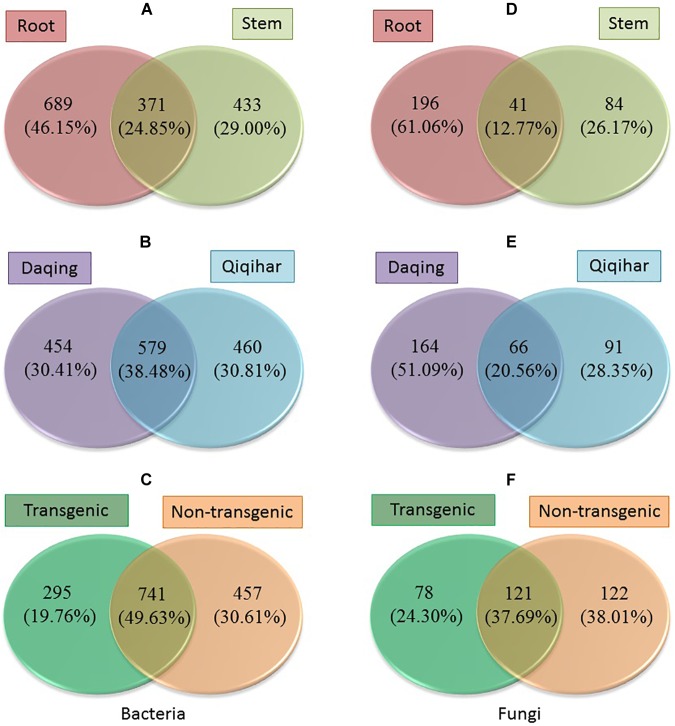
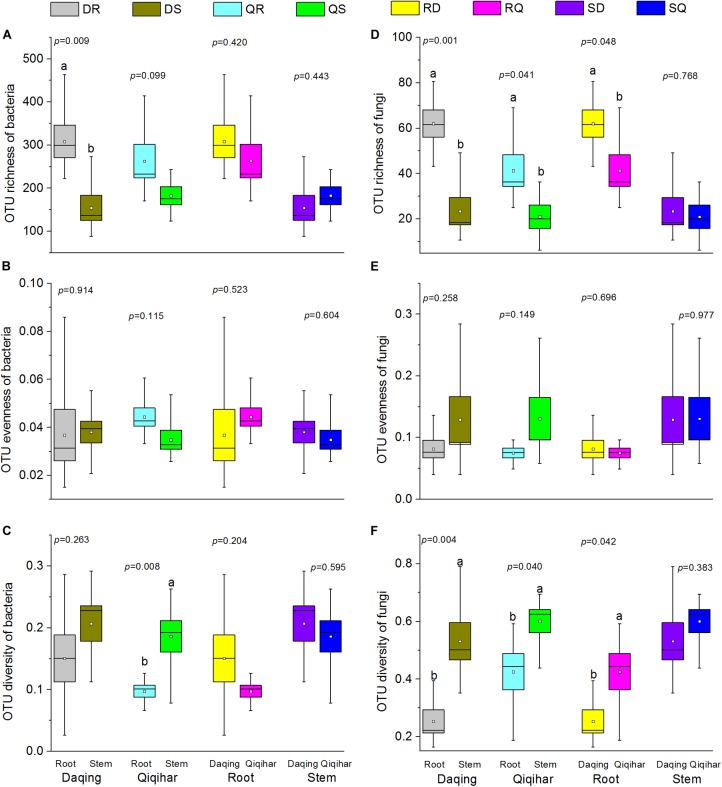
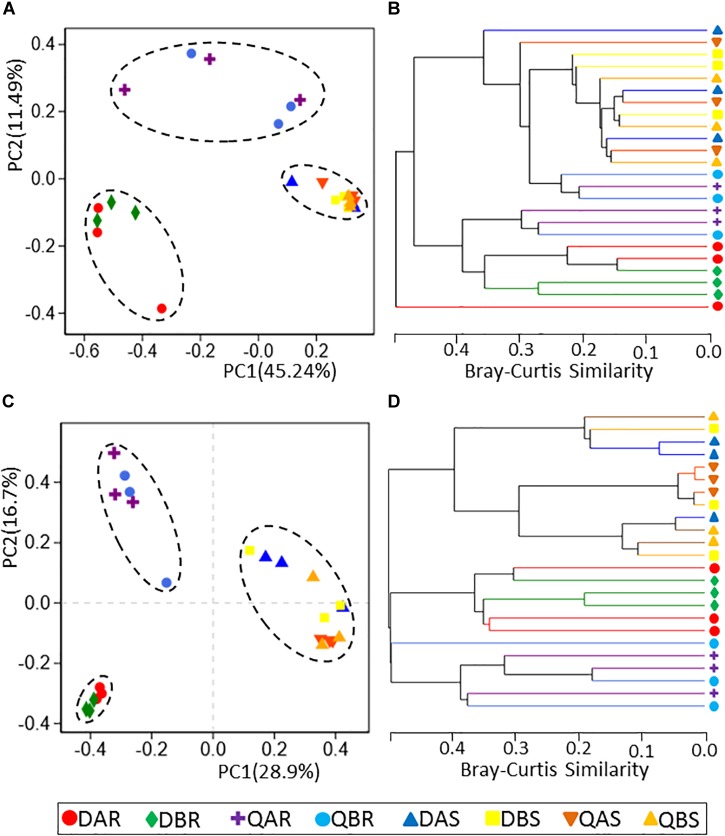
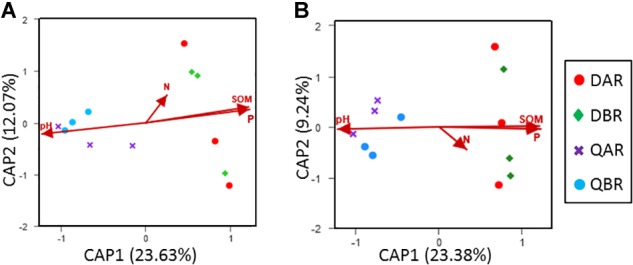
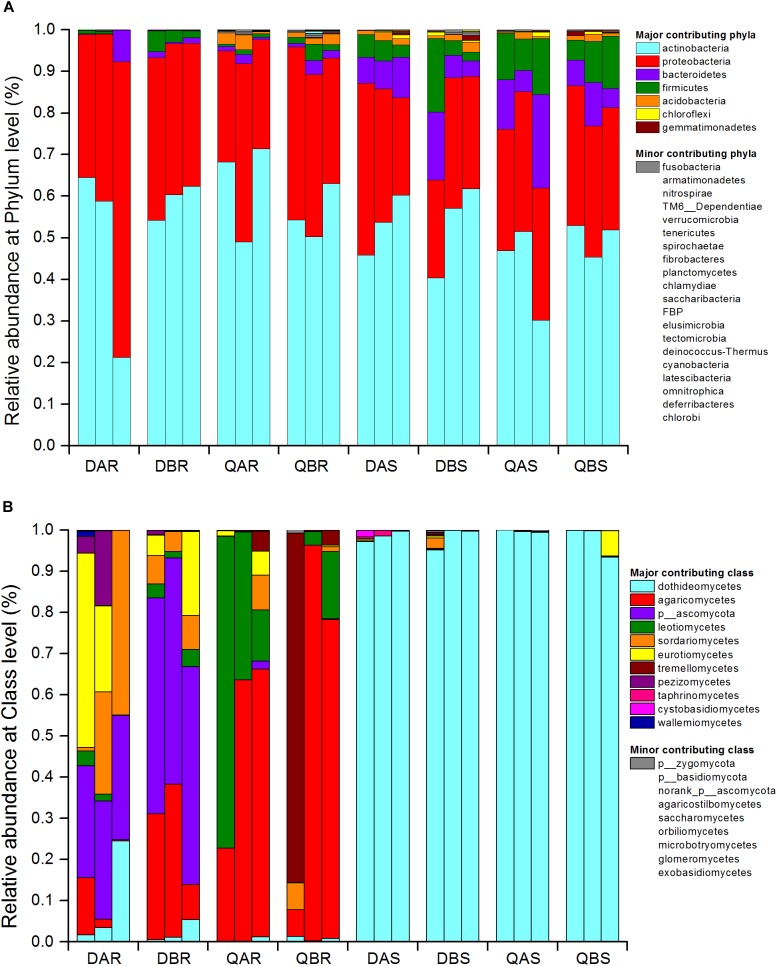
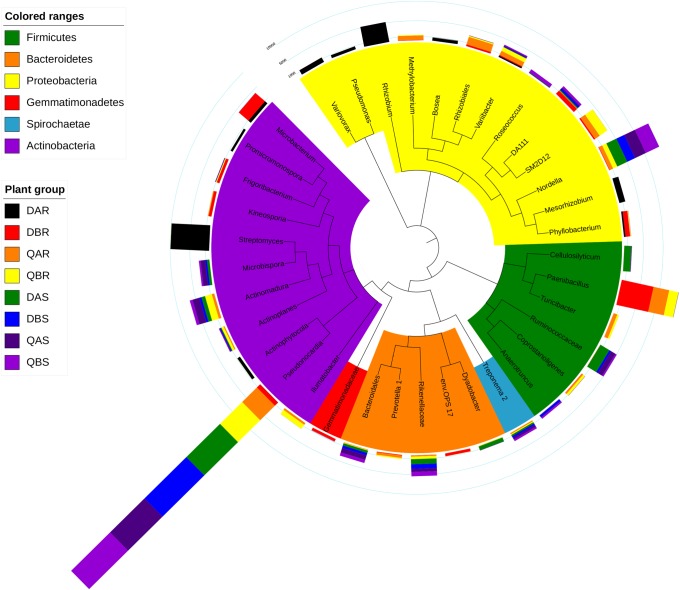
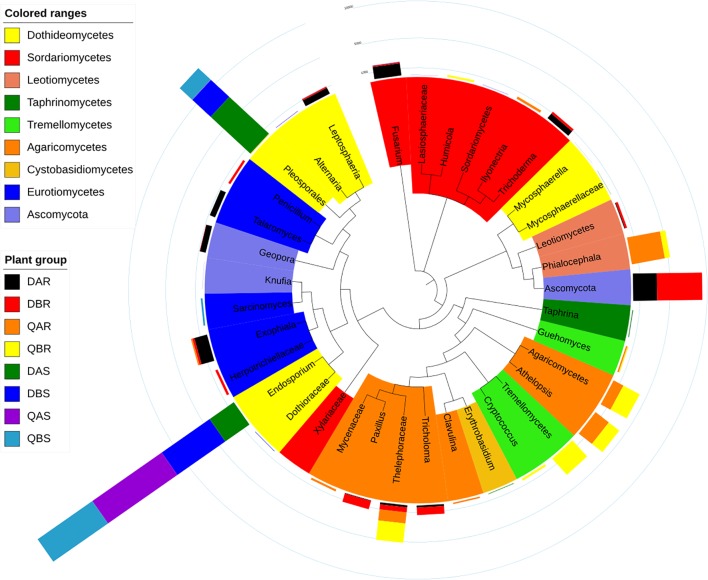
Microbial communities associated with plants represent key determinants of plant health, survival, and growth. However, a good understanding of the structural composition of the bacterial and fungal microbiome present in different plant tissues and growing environments, especially in transgenic woody plants, is required. In the present study, we hypothesized that environmental conditions, ecological niches, and transgenic events could influence the community structure of plant-associated microorganisms (bacterial and fungal endophytes). We sampled the root and stem endospheres of field-grown transgenic and non-transgenic poplar trees ( × ) and applied 16S rRNA and internal transcribed spacer amplicon Illumina MiSeq sequencing to determine the bacterial and fungal communities associated with the different plant habitats and tissues. We found that actinobacteria, proteobacteria, bacteroidetes, and firmicutes were the dominant endophytic bacteria, and the fungal community was dominated by dothideomycetes, agaricomycetes, leotiomycetes, and sordariomycetes. In conclusion, transgenic events did not affect the endophytic bacterial and fungal diversity of poplar trees. The bacterial and fungal community structure depends on the pH and the soil organic matter content. Each plant tissue represents a unique ecological niche for the microbial communities. Finally, we identified the indicator operational taxonomic units (OTUs) and core microbiome associated with the different plant tissues of and different environmental conditions. The results provide a basis for further study of host-microbial interactions with the identified abundant OTUs of .
与植物相关的微生物群落是植物健康、存活和生长的关键决定因素。然而,需要深入了解不同植物组织和生长环境(特别是转基因木本植物)中细菌和真菌微生物组的结构组成。在本研究中,我们假设环境条件、生态位和转基因事件会影响植物相关微生物(细菌和真菌内生菌)的群落结构。我们对田间种植的转基因和非转基因杨树(×)的根和茎内圈进行了采样,并应用16S rRNA和内部转录间隔区扩增子Illumina MiSeq测序来确定与不同植物生境和组织相关的细菌和真菌群落。我们发现放线菌、变形菌、拟杆菌和厚壁菌是主要的内生细菌,真菌群落则以座囊菌纲、伞菌纲、柔膜菌纲和粪壳菌纲为主。总之,转基因事件并未影响杨树的内生细菌和真菌多样性。细菌和真菌群落结构取决于pH值和土壤有机质含量。每个植物组织都代表着微生物群落的一个独特生态位。最后,我们确定了与杨树不同植物组织和不同环境条件相关的指示性可操作分类单元(OTU)和核心微生物组。研究结果为进一步研究宿主与微生物的相互作用以及已鉴定出的丰富OTU提供了基础。