Wang Sheng, Sun Siqi, Li Zhen, Zhang Renyu, Xu Jinbo
Toyota Technological Institute at Chicago, Chicago, Illinois, United States of America.
PLoS Comput Biol. 2017 Jan 5;13(1):e1005324. doi: 10.1371/journal.pcbi.1005324. eCollection 2017 Jan.
Protein contacts contain key information for the understanding of protein structure and function and thus, contact prediction from sequence is an important problem. Recently exciting progress has been made on this problem, but the predicted contacts for proteins without many sequence homologs is still of low quality and not very useful for de novo structure prediction.
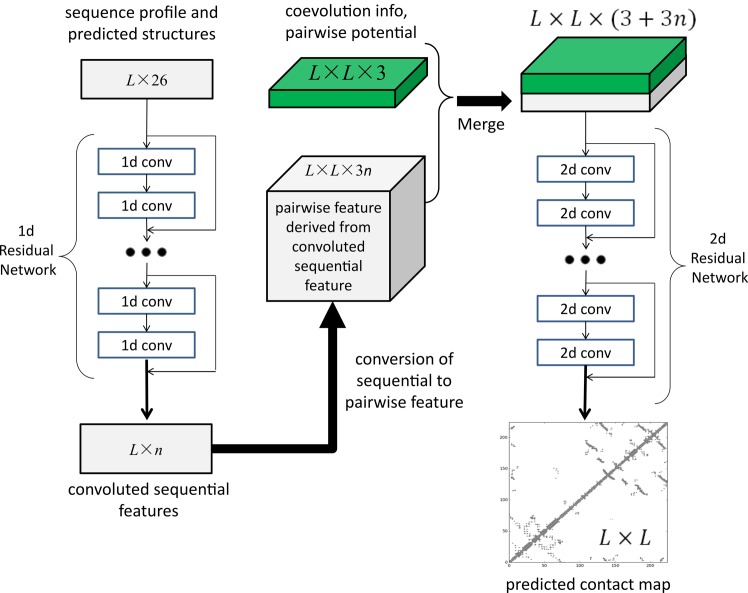
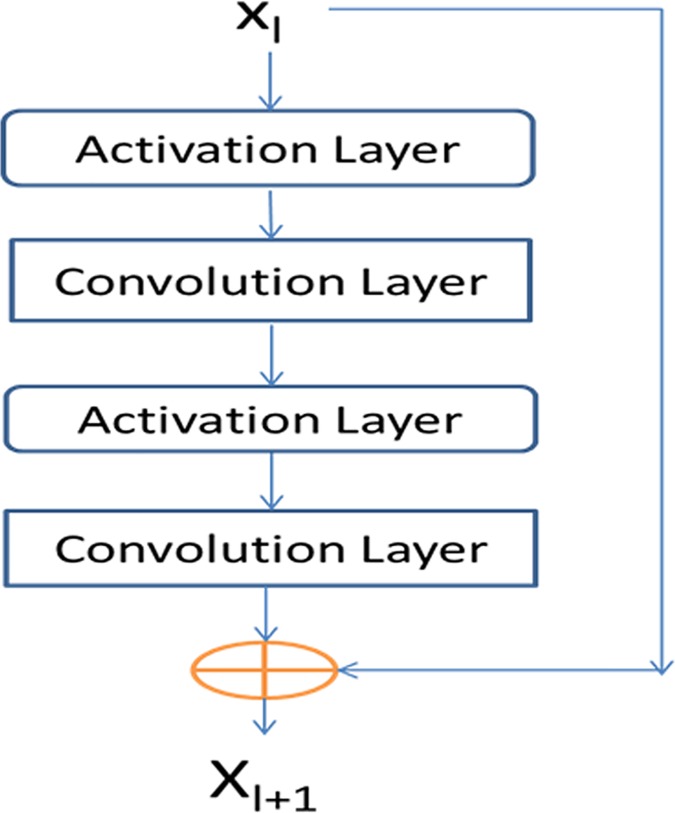
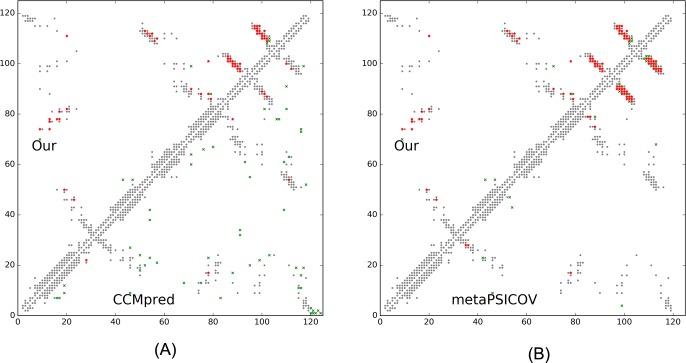
This paper presents a new deep learning method that predicts contacts by integrating both evolutionary coupling (EC) and sequence conservation information through an ultra-deep neural network formed by two deep residual neural networks. The first residual network conducts a series of 1-dimensional convolutional transformation of sequential features; the second residual network conducts a series of 2-dimensional convolutional transformation of pairwise information including output of the first residual network, EC information and pairwise potential. By using very deep residual networks, we can accurately model contact occurrence patterns and complex sequence-structure relationship and thus, obtain higher-quality contact prediction regardless of how many sequence homologs are available for proteins in question.
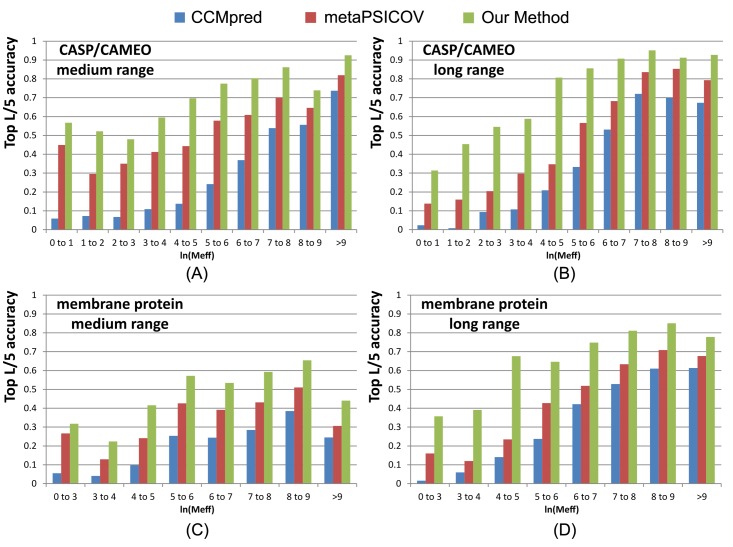
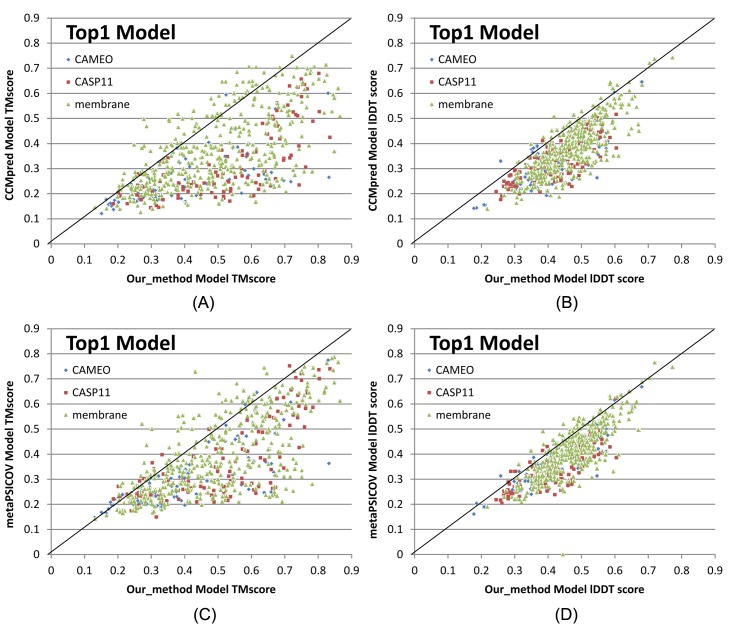
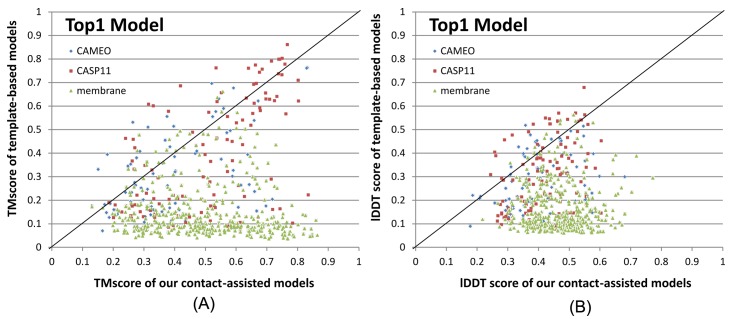
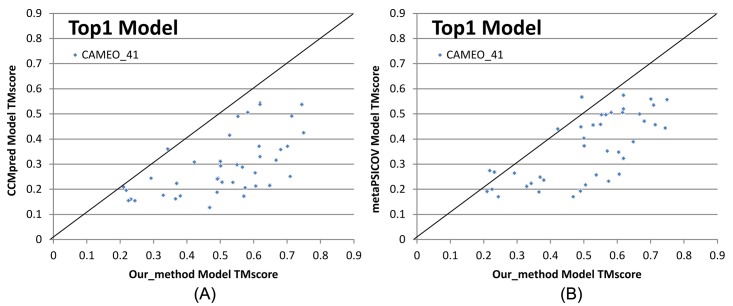
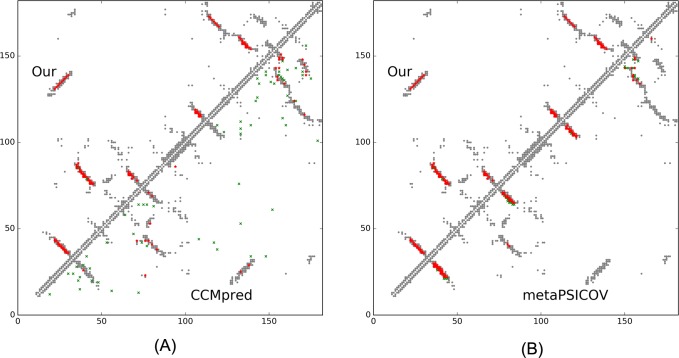
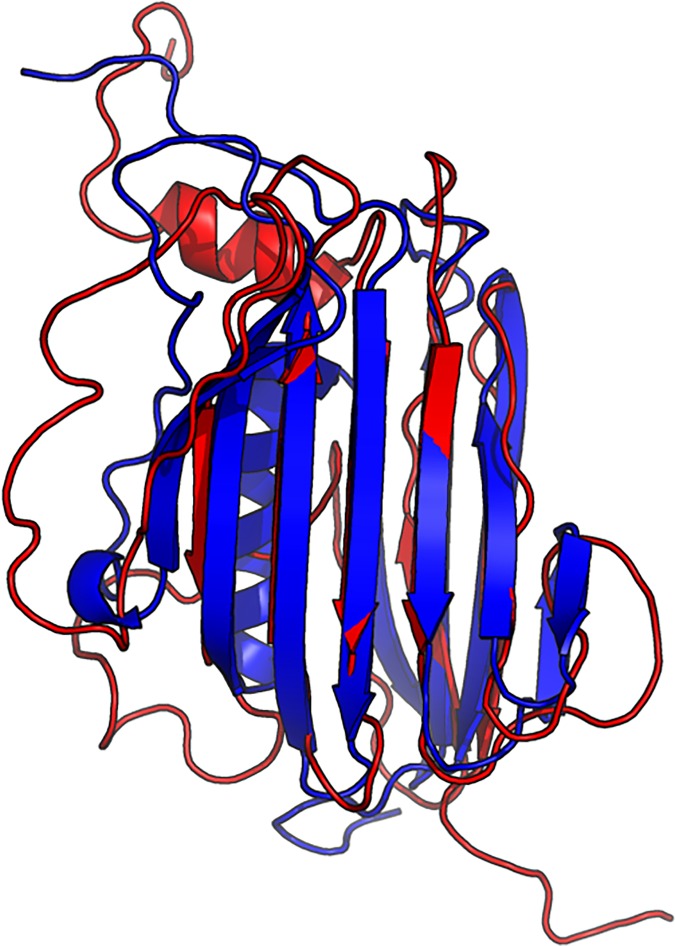
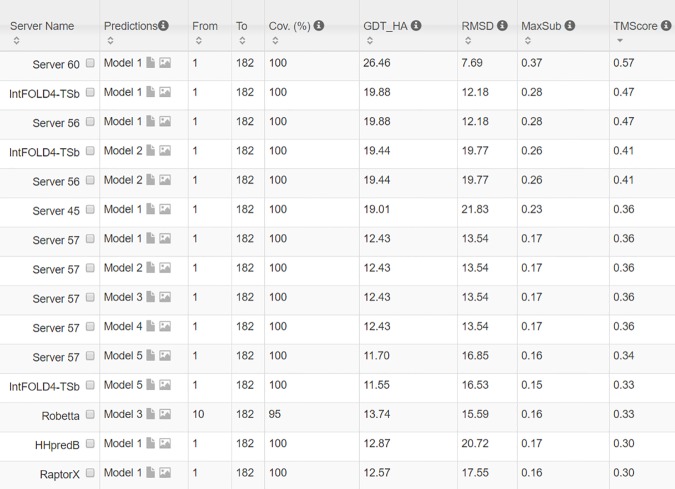
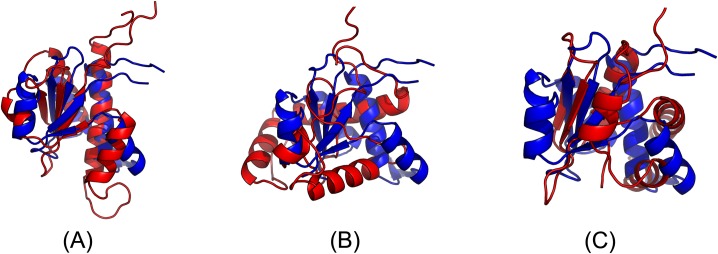
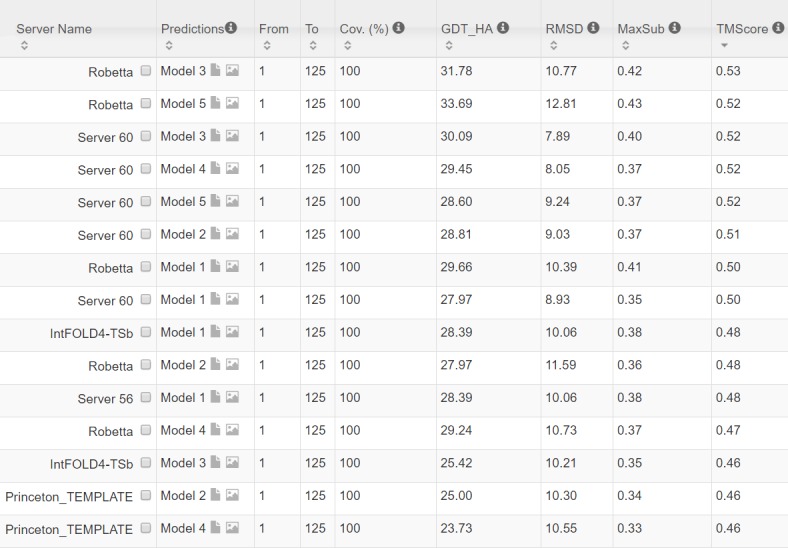
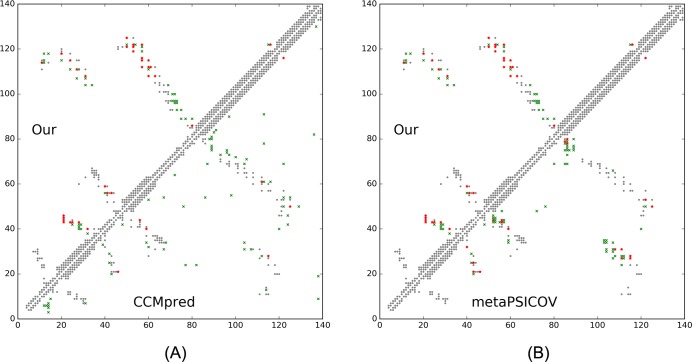
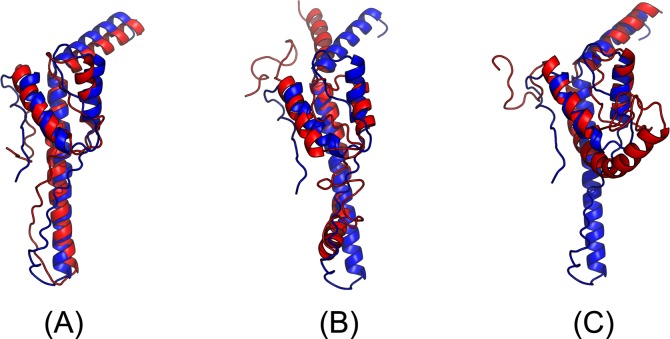
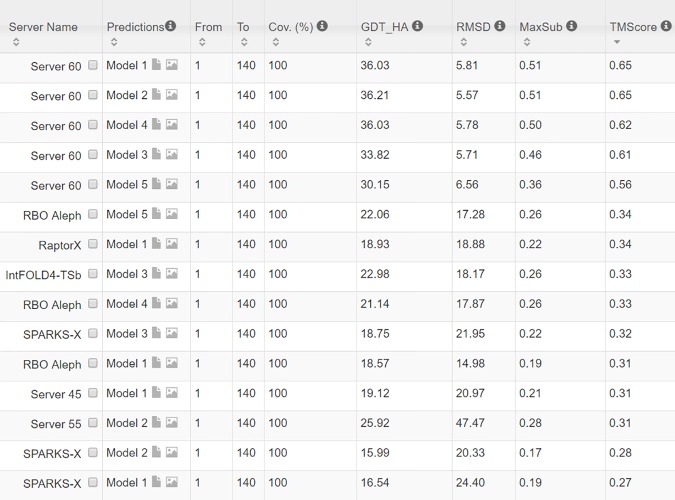
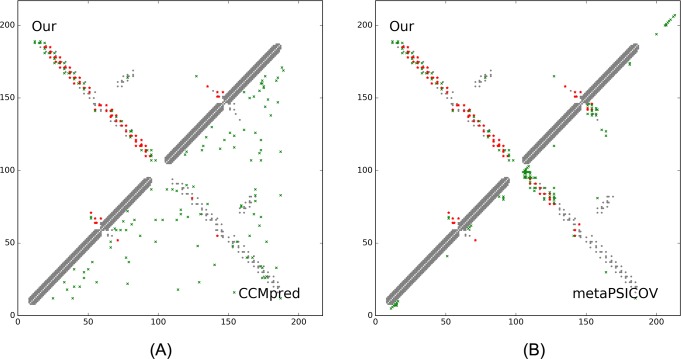
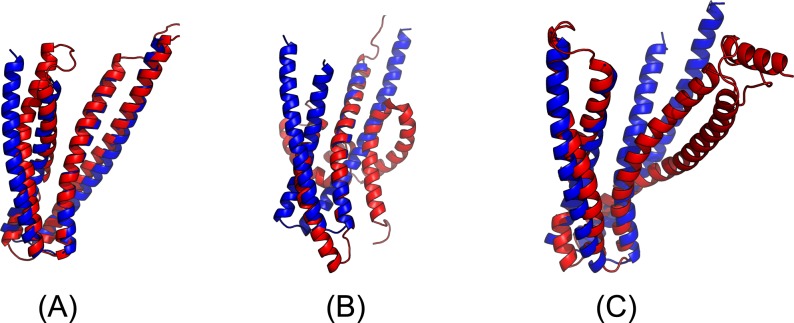
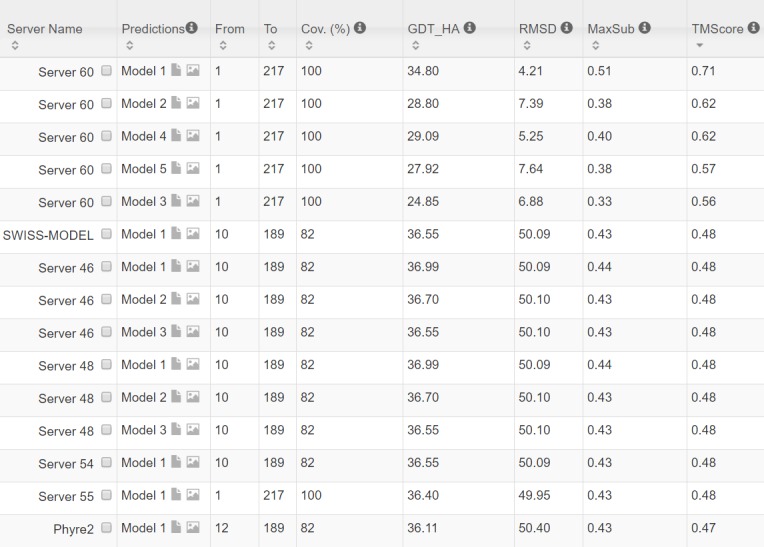
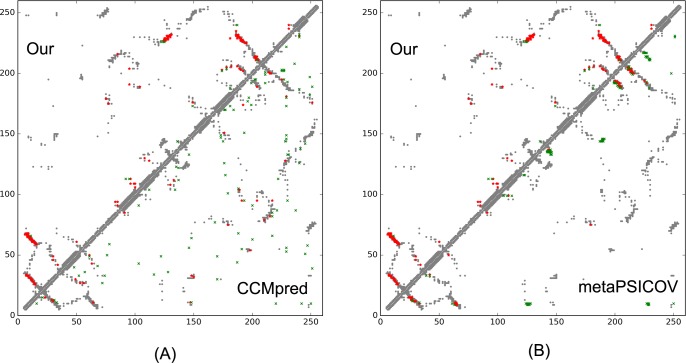
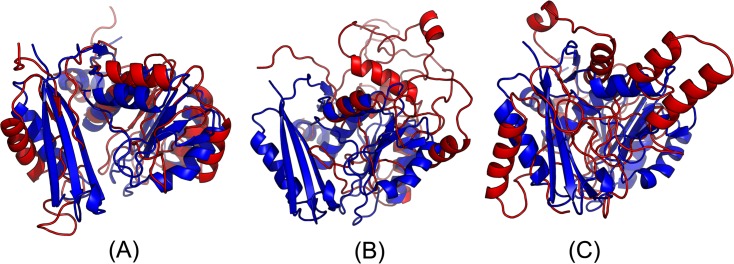
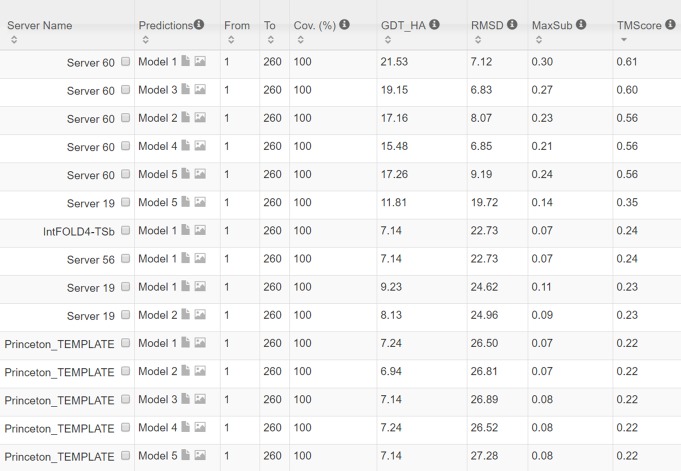
Our method greatly outperforms existing methods and leads to much more accurate contact-assisted folding. Tested on 105 CASP11 targets, 76 past CAMEO hard targets, and 398 membrane proteins, the average top L long-range prediction accuracy obtained by our method, one representative EC method CCMpred and the CASP11 winner MetaPSICOV is 0.47, 0.21 and 0.30, respectively; the average top L/10 long-range accuracy of our method, CCMpred and MetaPSICOV is 0.77, 0.47 and 0.59, respectively. Ab initio folding using our predicted contacts as restraints but without any force fields can yield correct folds (i.e., TMscore>0.6) for 203 of the 579 test proteins, while that using MetaPSICOV- and CCMpred-predicted contacts can do so for only 79 and 62 of them, respectively. Our contact-assisted models also have much better quality than template-based models especially for membrane proteins. The 3D models built from our contact prediction have TMscore>0.5 for 208 of the 398 membrane proteins, while those from homology modeling have TMscore>0.5 for only 10 of them. Further, even if trained mostly by soluble proteins, our deep learning method works very well on membrane proteins. In the recent blind CAMEO benchmark, our fully-automated web server implementing this method successfully folded 6 targets with a new fold and only 0.3L-2.3L effective sequence homologs, including one β protein of 182 residues, one α+β protein of 125 residues, one α protein of 140 residues, one α protein of 217 residues, one α/β of 260 residues and one α protein of 462 residues. Our method also achieved the highest F1 score on free-modeling targets in the latest CASP (Critical Assessment of Structure Prediction), although it was not fully implemented back then.
蛋白质接触包含理解蛋白质结构和功能的关键信息,因此,从序列预测接触是一个重要问题。最近在这个问题上取得了令人兴奋的进展,但对于没有许多序列同源物的蛋白质,预测的接触质量仍然很低,对从头结构预测不太有用。
本文提出了一种新的深度学习方法,该方法通过由两个深度残差神经网络组成的超深度神经网络整合进化耦合(EC)和序列保守信息来预测接触。第一个残差网络对序列特征进行一系列一维卷积变换;第二个残差网络对包括第一个残差网络的输出、EC信息和成对势在内的成对信息进行一系列二维卷积变换。通过使用非常深的残差网络,我们可以准确地对接触出现模式和复杂的序列-结构关系进行建模,从而无论所讨论的蛋白质有多少序列同源物,都能获得更高质量的接触预测。
我们的方法大大优于现有方法,并导致更准确的接触辅助折叠。在105个CASP11目标、76个过去的CAMEO硬目标和398个膜蛋白上进行测试,我们的方法、一种代表性的EC方法CCMpred和CASP11获胜者MetaPSICOV获得的平均前L个长程预测准确率分别为0.47、0.21和0.30;我们的方法、CCMpred和MetaPSICOV的平均前L/10长程准确率分别为0.77、0.47和0.59。使用我们预测的接触作为约束但不使用任何力场的从头折叠可以为579个测试蛋白中的203个产生正确的折叠(即TMscore>0.6),而使用MetaPSICOV和CCMpred预测的接触分别只能为其中的79个和62个产生正确折叠。我们的接触辅助模型的质量也比基于模板的模型好得多,特别是对于膜蛋白。从我们的接触预测构建的3D模型在398个膜蛋白中有208个的TMscore>0.5,而同源建模构建的3D模型只有10个的TMscore>0.5。此外,即使主要由可溶性蛋白训练,我们的深度学习方法在膜蛋白上也表现得非常好。在最近的盲CAMEO基准测试中,我们实现此方法的全自动网络服务器成功折叠了6个具有新折叠且有效序列同源物仅为0.3L - 2.3L的目标,包括一个182个残基的β蛋白、一个125个残基的α + β蛋白、一个140个残基的α蛋白、一个217个残基的α蛋白、一个260个残基的α/β蛋白和一个462个残基的α蛋白。我们的方法在最新的CASP(结构预测关键评估)中的自由建模目标上也获得了最高的F1分数,尽管当时它没有完全实现。