Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan, USA.
Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan, USA; Department of Biological Chemistry, University of Michigan, Ann Arbor, Michigan, USA.
J Biol Chem. 2021 Jul;297(1):100870. doi: 10.1016/j.jbc.2021.100870. Epub 2021 Jun 11.
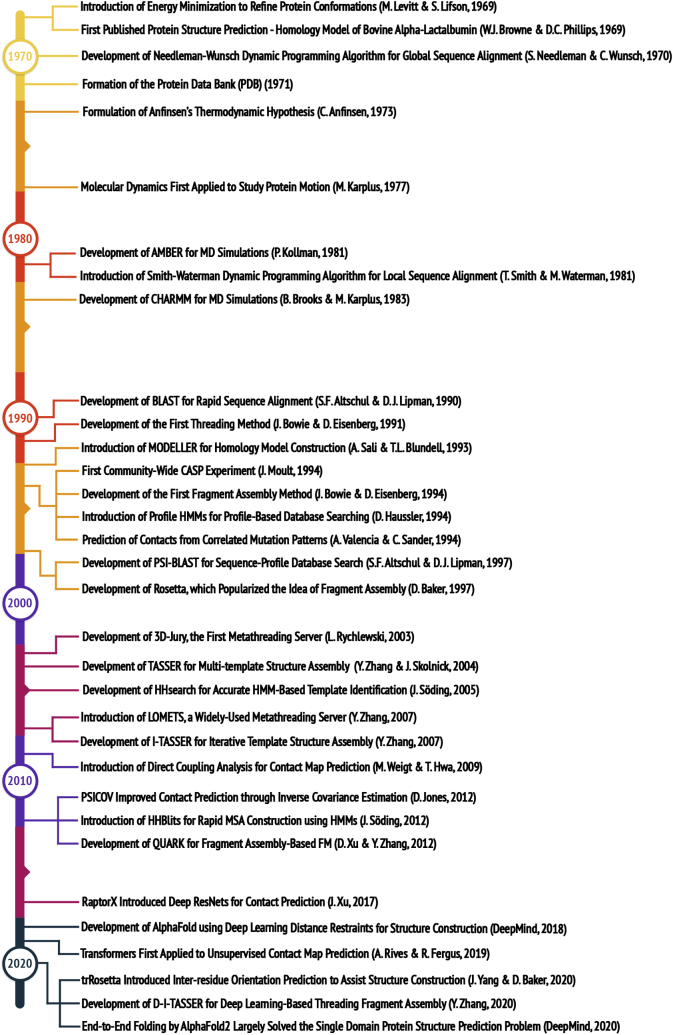
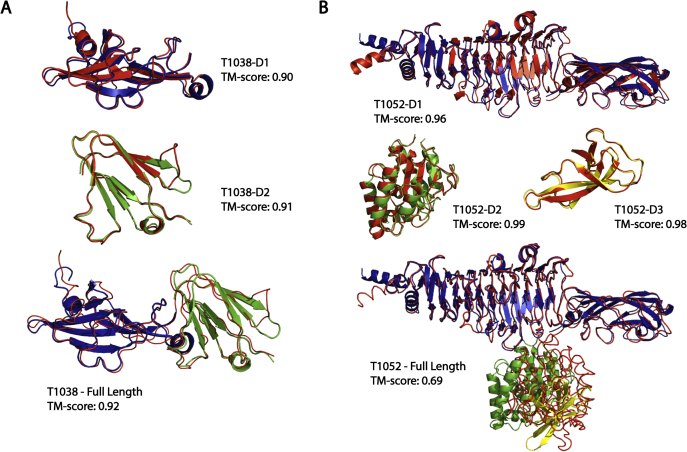
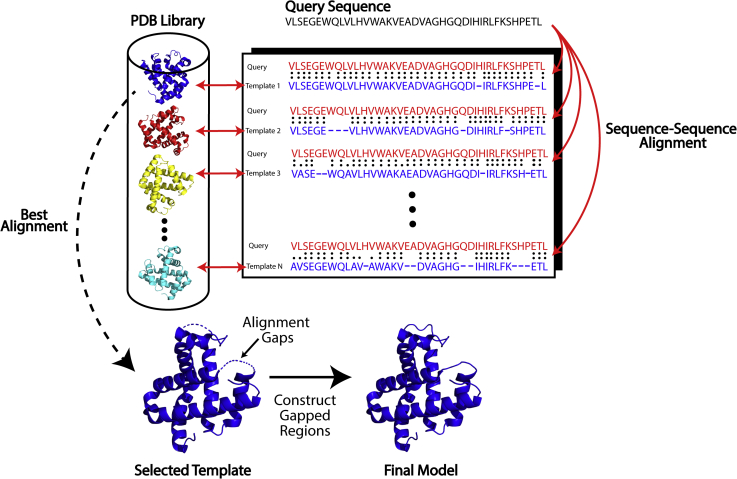
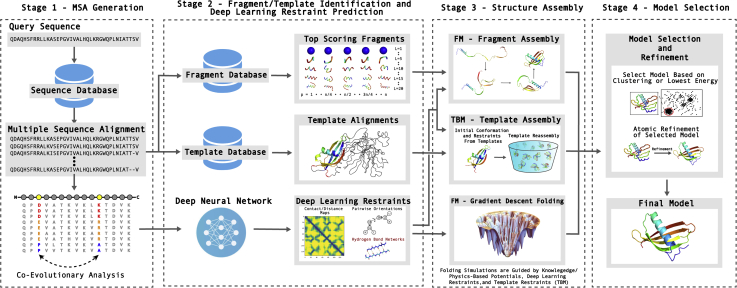
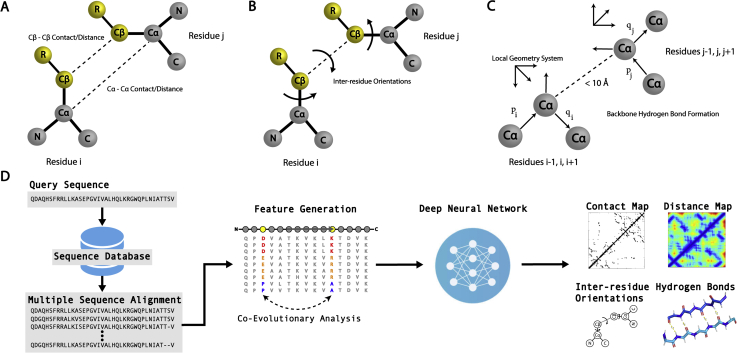
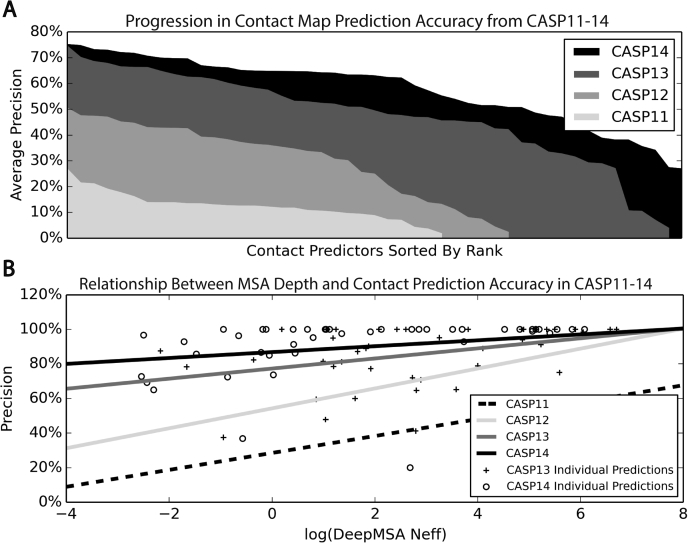
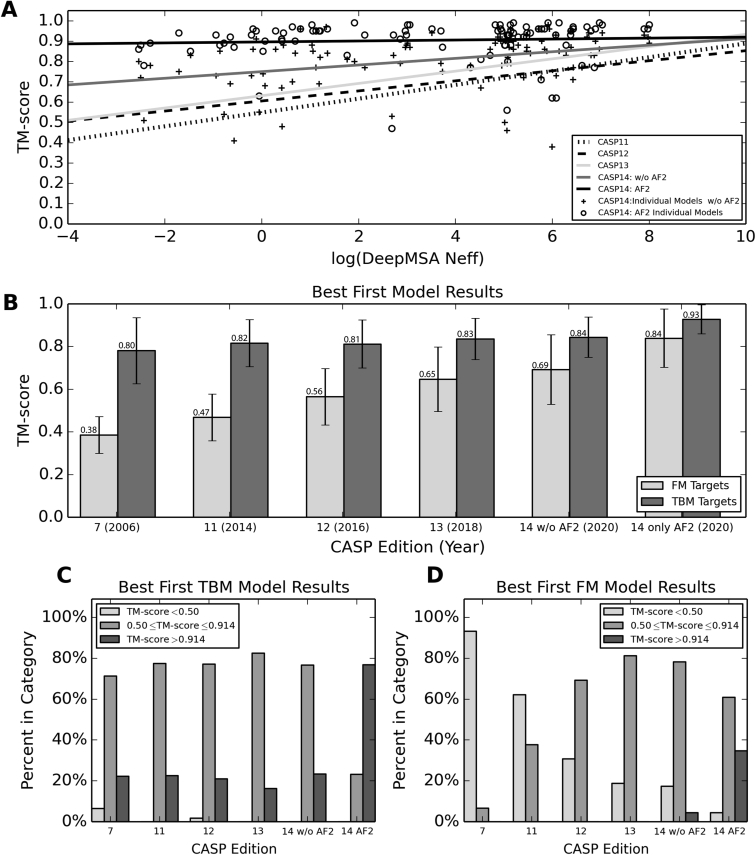
Since Anfinsen demonstrated that the information encoded in a protein's amino acid sequence determines its structure in 1973, solving the protein structure prediction problem has been the Holy Grail of structural biology. The goal of protein structure prediction approaches is to utilize computational modeling to determine the spatial location of every atom in a protein molecule starting from only its amino acid sequence. Depending on whether homologous structures can be found in the Protein Data Bank (PDB), structure prediction methods have been historically categorized as template-based modeling (TBM) or template-free modeling (FM) approaches. Until recently, TBM has been the most reliable approach to predicting protein structures, and in the absence of reliable templates, the modeling accuracy sharply declines. Nevertheless, the results of the most recent community-wide assessment of protein structure prediction experiment (CASP14) have demonstrated that the protein structure prediction problem can be largely solved through the use of end-to-end deep machine learning techniques, where correct folds could be built for nearly all single-domain proteins without using the PDB templates. Critically, the model quality exhibited little correlation with the quality of available template structures, as well as the number of sequence homologs detected for a given target protein. Thus, the implementation of deep-learning techniques has essentially broken through the 50-year-old modeling border between TBM and FM approaches and has made the success of high-resolution structure prediction significantly less dependent on template availability in the PDB library.
自 Anfinsen 于 1973 年证明蛋白质的氨基酸序列中所编码的信息决定其结构以来,解决蛋白质结构预测问题一直是结构生物学的圣杯。蛋白质结构预测方法的目标是利用计算建模,从蛋白质分子的氨基酸序列出发,确定其每个原子的空间位置。根据是否可以在蛋白质数据库(PDB)中找到同源结构,结构预测方法在历史上被分为基于模板的建模(TBM)或无模板建模(FM)方法。直到最近,TBM 一直是预测蛋白质结构最可靠的方法,而且在缺乏可靠模板的情况下,建模准确性急剧下降。然而,最近的蛋白质结构预测实验(CASP14)的社区评估结果表明,通过使用端到端的深度学习技术,蛋白质结构预测问题可以得到很大程度的解决,几乎所有的单一结构域蛋白质都可以在不使用 PDB 模板的情况下构建正确的折叠。关键的是,模型质量与可用模板结构的质量以及为给定目标蛋白质检测到的序列同源物的数量几乎没有相关性。因此,深度学习技术的实施基本上打破了 TBM 和 FM 方法之间 50 年的建模边界,使得高分辨率结构预测的成功不再严重依赖于 PDB 库中的模板可用性。