Charboneau David J, Brudvig Gary W, Hazari Nilay, Lant Hannah M C, Saydjari Andrew K
Department of Chemistry, Yale University, P.O. Box 208107, New Haven, Connecticut 06520, United States.
ACS Catal. 2019 Apr 5;9(4):3228-3241. doi: 10.1021/acscatal.9b00566. Epub 2019 Mar 14.
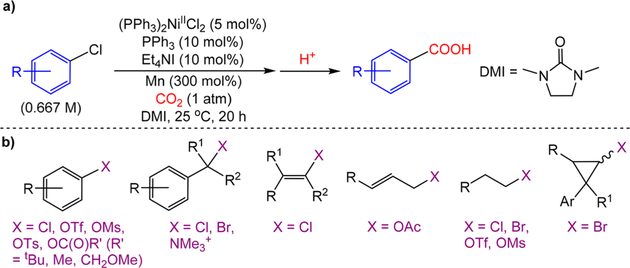
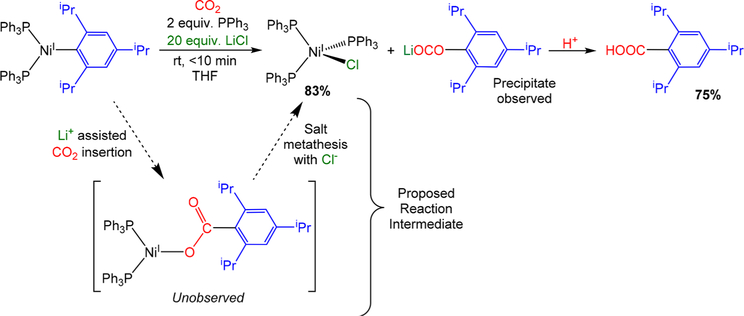
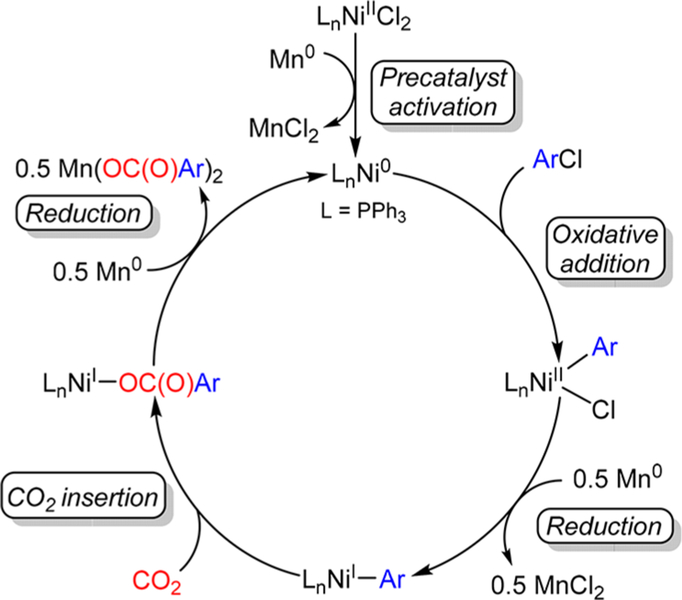
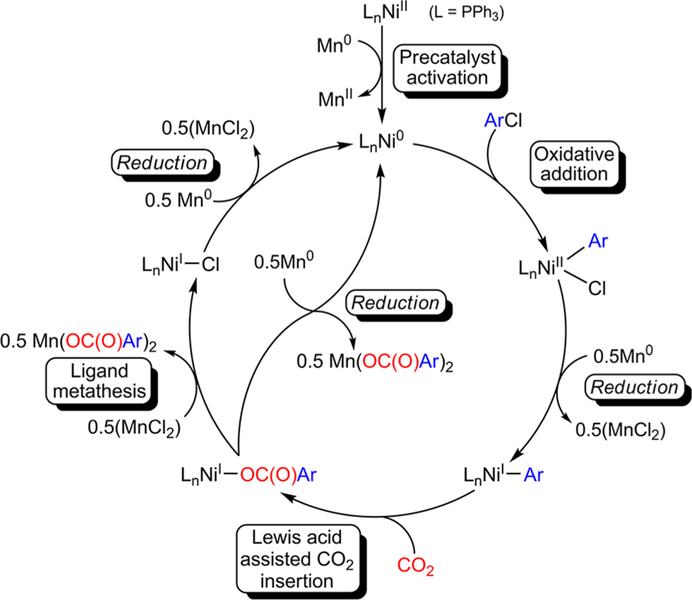
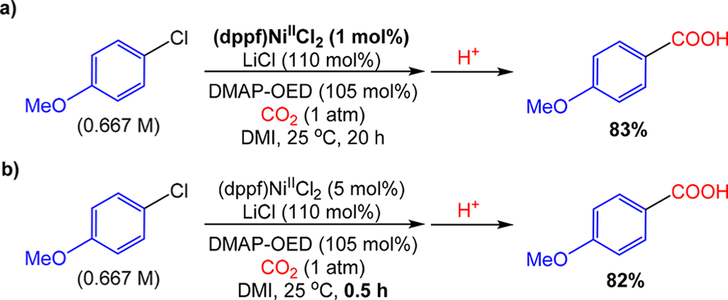
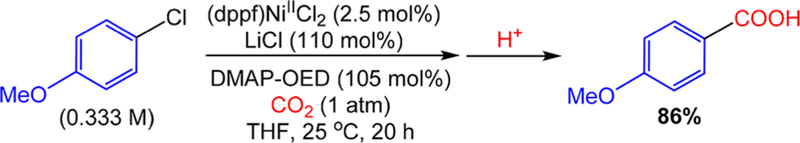
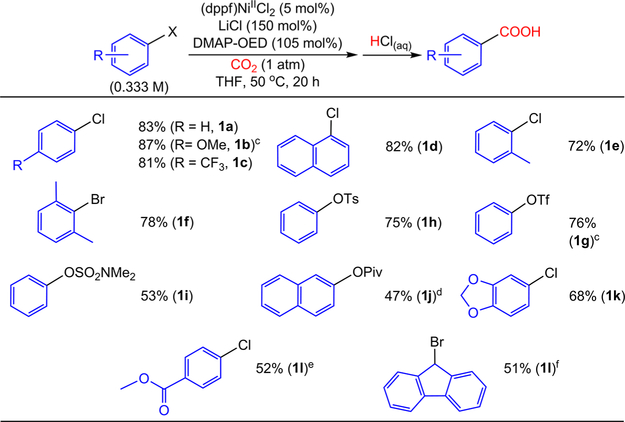
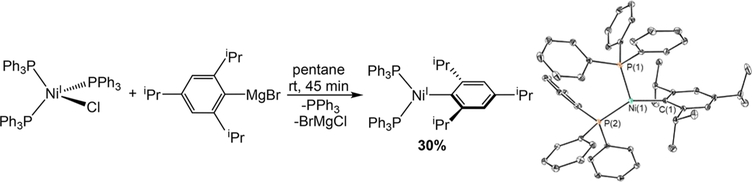
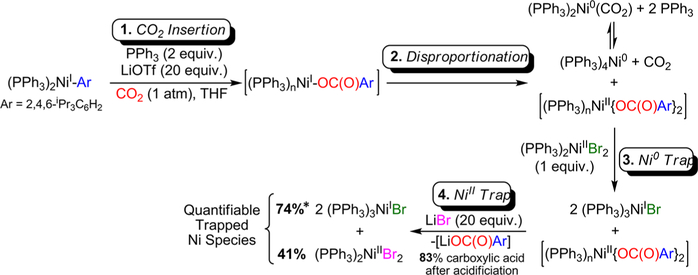
The nickel-catalyzed carboxylation of organic halides or pseudohalides using carbon dioxide is an emerging method to prepare synthetically valuable carboxylic acids. Here, we report a detailed mechanistic investigation of these reactions using the carboxylation of aryl halides with (PPh)NiCl as a model reaction. Our studies allow us to understand several general features of nickel-catalyzed carboxylation reactions. For example, we demonstrate that both a Lewis acid and halide source are beneficial for catalysis. To this end, we establish that heterogeneous Mn(0) and Zn(0) reductants are multifaceted reagents that generate noninnocent Mn(II) or Zn(II) Lewis acids upon oxidation. In a key result, a rare example of a well-defined nickel(I) aryl complex is isolated, and it is demonstrated that its reaction with carbon dioxide results in the formation of a carboxylic acid in high yield (after workup). The carbon dioxide insertion product undergoes rapid decomposition, which ca These three oxidation states correspond to the onbe circumvented by a ligand metathesis reaction with a halide source. Our studies have led to both a revised mechanism and the development of a broadly applicable strategy to improve reductive carboxylation reactions. A critical component of this strategy is that we have replaced the heterogeneous Mn(0) reductant typically used in catalysis with a well-defined homogeneous organic reductant. Through its use, we have increased the range of ancillary ligands, additives, and substrates that are compatible with the reaction. This has enabled us to perform reductive carboxylations at low catalyst loadings. Additionally, we demonstrate that reductive carboxylations of organic (pseudo)halides can be achieved in high yields in more practically useful, non-amide solvents. Our results describe a mechanistically guided strategy to improve reductive carboxylations through the use of a homogeneous organic reductant, which may be broadly translatable to a wide range of cross-electrophile coupling reactions.
使用二氧化碳对有机卤化物或拟卤化物进行镍催化的羧化反应是一种新兴的制备具有合成价值的羧酸的方法。在此,我们报道了以芳基卤化物与(PPh)NiCl的羧化反应为模型反应,对这些反应进行的详细机理研究。我们的研究使我们能够了解镍催化羧化反应的几个一般特征。例如,我们证明路易斯酸和卤化物源都有利于催化反应。为此,我们确定多相的Mn(0)和Zn(0)还原剂是多面试剂,它们在氧化后会生成非惰性的Mn(II)或Zn(II)路易斯酸。在一个关键结果中,分离出了一个明确的镍(I)芳基配合物的罕见例子,并且证明它与二氧化碳反应能高产率地生成羧酸(后处理后)。二氧化碳插入产物会迅速分解,这可以通过与卤化物源的配体复分解反应来避免。我们的研究既导致了机理的修订,也开发了一种广泛适用的策略来改进还原羧化反应。该策略的一个关键组成部分是,我们用一种明确的均相有机还原剂取代了催化中通常使用的多相Mn(0)还原剂。通过使用这种还原剂,我们扩大了与反应兼容的辅助配体、添加剂和底物的范围。这使我们能够在低催化剂负载量下进行还原羧化反应。此外,我们证明在更实用的非酰胺溶剂中,有机(拟)卤化物的还原羧化反应可以高产率实现。我们的结果描述了一种通过使用均相有机还原剂来改进还原羧化反应的机理指导策略,该策略可能广泛适用于各种交叉亲电偶联反应。