Computer Science and Engineering Technology, University of Houston-Downtown, Houston, TX 77002, USA.
Biology and Biochemistry, University of Houston, Houston, TX 77204, USA.
Nucleic Acids Res. 2019 Jul 26;47(13):e78. doi: 10.1093/nar/gkz315.
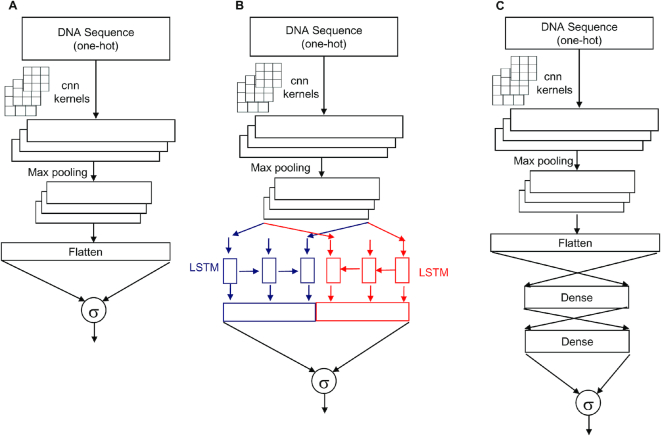
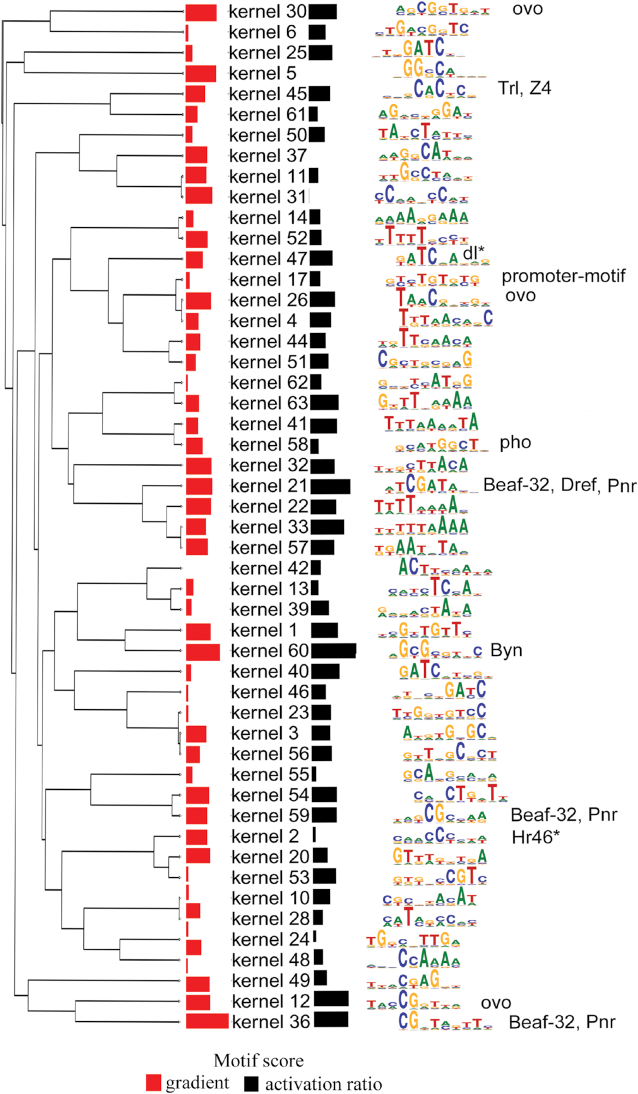
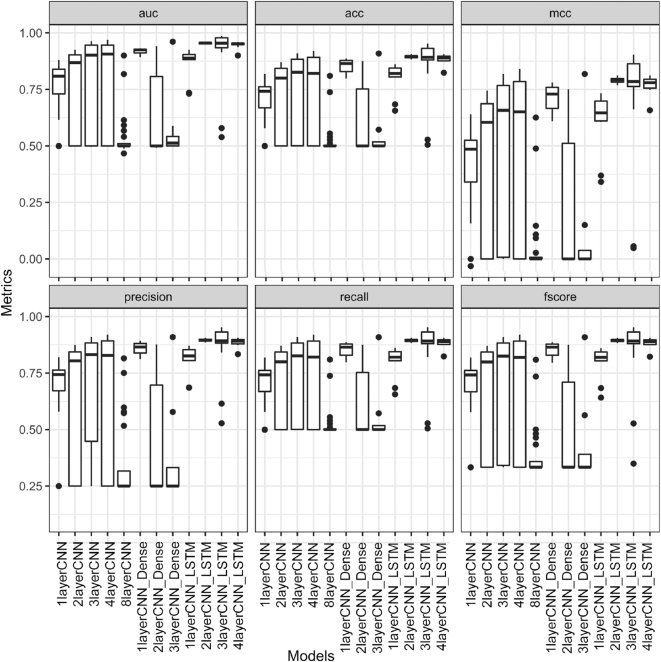
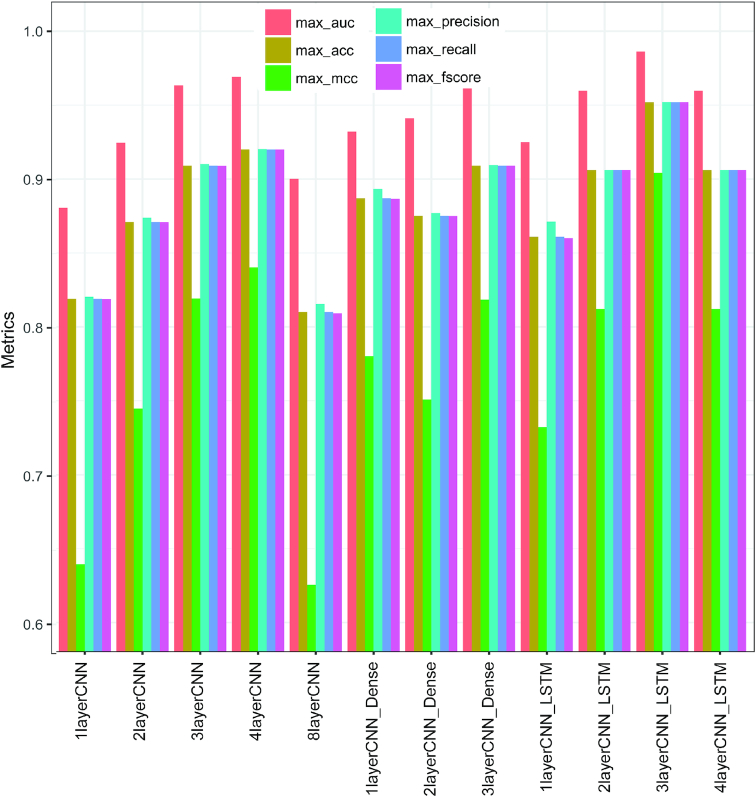
Genomes are organized into self-interacting chromatin regions called topologically associated domains (TADs). A significant number of TAD boundaries are shared across multiple cell types and conserved across species. Disruption of TAD boundaries may affect the expression of nearby genes and could lead to several diseases. Even though detection of TAD boundaries is important and useful, there are experimental challenges in obtaining high resolution TAD locations. Here, we present computational prediction of TAD boundaries from high resolution Hi-C data in fruit flies. By extensive exploration and testing of several deep learning model architectures with hyperparameter optimization, we show that a unique deep learning model consisting of three convolution layers followed by a long short-term-memory layer achieves an accuracy of 96%. This outperforms feature-based models' accuracy of 91% and an existing method's accuracy of 73-78% based on motif TRAP scores. Our method also detects previously reported motifs such as Beaf-32 that are enriched in TAD boundaries in fruit flies and also several unreported motifs.
基因组组织成称为拓扑关联域(TAD)的自我相互作用染色质区域。大量的 TAD 边界在多种细胞类型中共享,并在物种间保守。TAD 边界的破坏可能会影响附近基因的表达,并可能导致多种疾病。尽管 TAD 边界的检测很重要且有用,但在获得高分辨率 TAD 位置方面存在实验挑战。在这里,我们提出了一种从果蝇高分辨率 Hi-C 数据中预测 TAD 边界的计算方法。通过对几种深度学习模型结构进行广泛的探索和测试,并进行超参数优化,我们表明,由三个卷积层和一个长短期记忆层组成的独特深度学习模型的准确率为 96%。这优于基于特征的模型的准确率 91%,以及基于 motif TRAP 分数的现有方法的准确率 73-78%。我们的方法还检测到了先前报道的果蝇 TAD 边界中富集的 Beaf-32 等motif,以及一些未报道的 motif。