Mores Kendall L, Cummins Benjamin R, Cassell Robert J, van Rijn Richard M
Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, West Lafayette, IN, United States.
Department of Chemistry, College of Science, West Lafayette, IN, United States.
Front Pharmacol. 2019 Apr 17;10:407. doi: 10.3389/fphar.2019.00407. eCollection 2019.
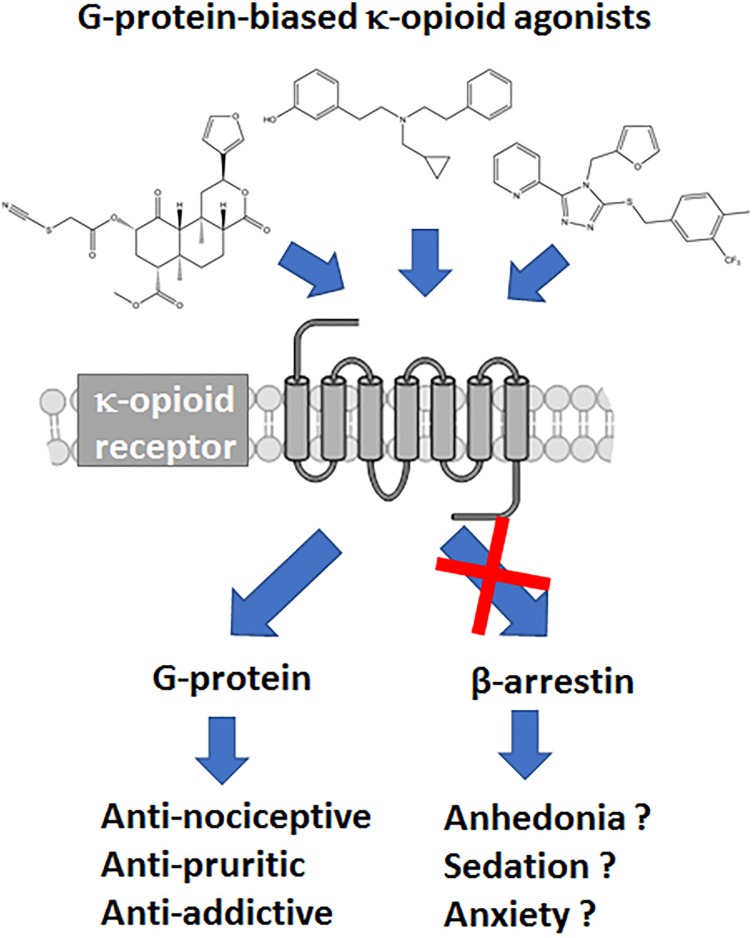
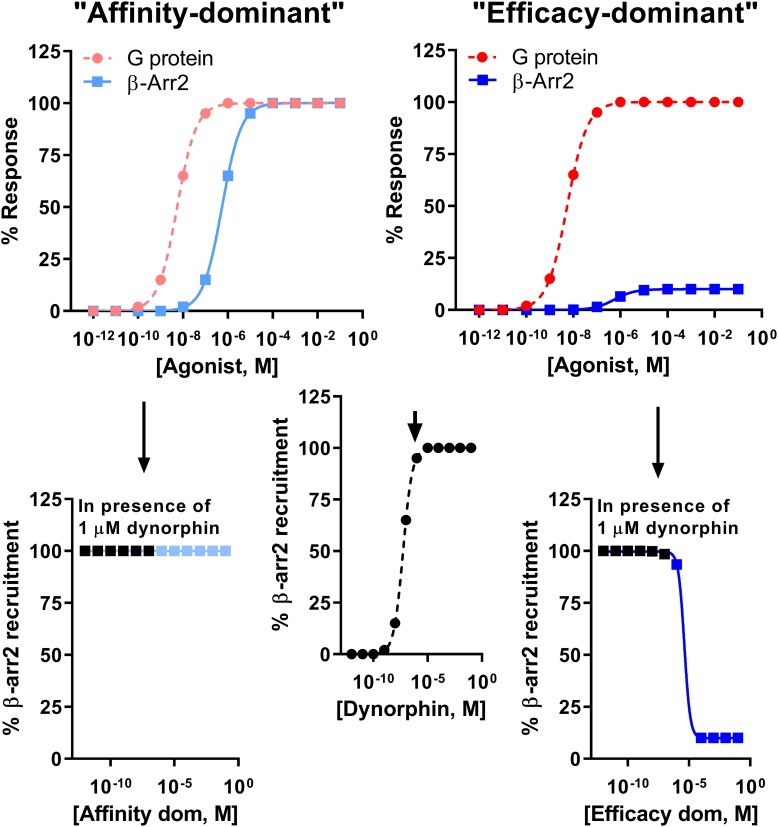
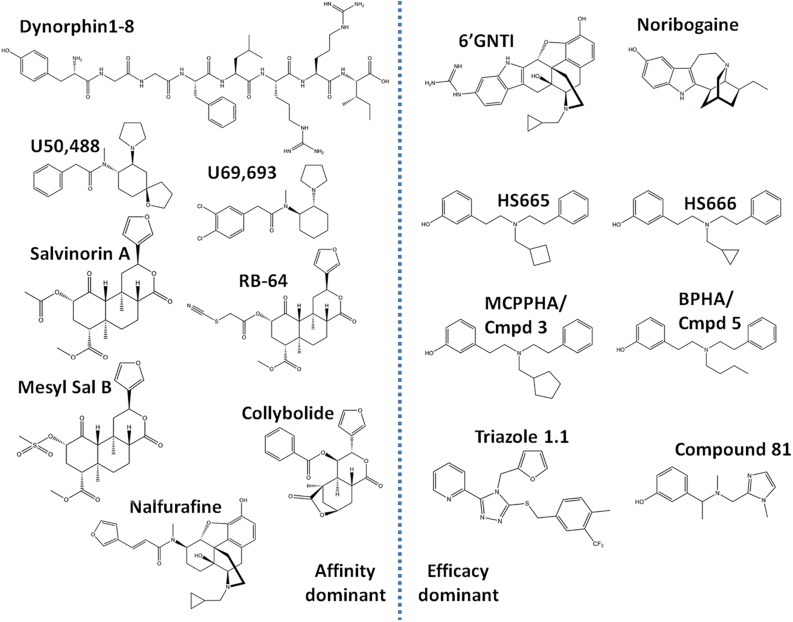
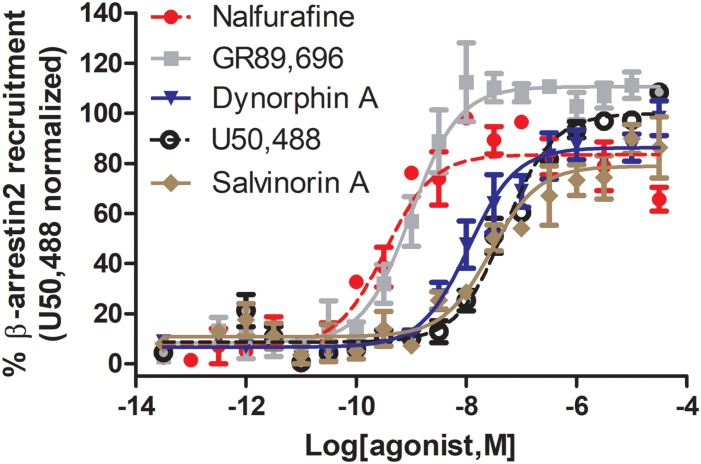
Between 2000 and 2005 several studies revealed that morphine is more potent and exhibits fewer side effects in beta-arrestin 2 knockout mice. These findings spurred efforts to develop opioids that signal primarily via G protein activation and do not, or only very weakly, recruit beta-arrestin. Development of such molecules targeting the mu opioid receptor initially outpaced those targeting the kappa, delta and nociceptin opioid receptors, with the G protein-biased mu opioid agonist oliceridine/TRV130 having completed phase III clinical trials with improved therapeutic window to treat moderate-to-severe acute pain. Recently however, there has been a sharp increase in the development of novel G protein-biased kappa agonists. It is hypothesized that G protein-biased kappa agonists can reduce pain and itch, but exhibit fewer side effects, such as anhedonia and psychosis, that have thus far limited the clinical development of unbiased kappa opioid agonists. Here we summarize recently discovered G protein-biased kappa agonists, comparing structures, degree of signal bias and preclinical effects. We specifically reviewed nalfurafine, 22-thiocyanatosalvinorin A (RB-64), mesyl-salvinorin B, 2-(4-(furan-2-ylmethyl)-5-((4-methyl-3-(trifluoromethyl)benzyl)thio)-4H-1,2,4-triazol-3-yl)pyridine (triazole 1.1), 3-(2-((cyclopropylmethyl)(phenethyl)amino)ethyl)phenol (HS666), -butyl--phenylethyl--3-hydroxyphenylethyl-amine (compound 5/BPHA), 6-guanidinonaltrindole (6'GNTI), and collybolide. These agonists encompass a variety of chemical scaffolds and range in both their potency and efficacy in terms of G protein signaling and beta-arrestin recruitment. Thus unsurprisingly, the behavioral responses reported for these agonists are not uniform. Yet, it is our conclusion that the kappa opioid field will benefit tremendously from future studies that compare several biased agonists and correlate the degree of signaling bias to a particular pharmacological response.
2000年至2005年间的多项研究表明,在β-抑制蛋白2基因敲除小鼠中,吗啡的效力更强且副作用更少。这些发现促使人们努力开发主要通过G蛋白激活发挥信号传导作用、不招募或仅非常微弱地招募β-抑制蛋白的阿片类药物。最初,针对μ阿片受体开发此类分子的进展超过了针对κ、δ和孤啡肽阿片受体的研究,G蛋白偏向性μ阿片激动剂oliceridine/TRV130已完成治疗中重度急性疼痛的III期临床试验,其治疗窗口有所改善。然而,最近新型G蛋白偏向性κ激动剂的开发急剧增加。据推测,G蛋白偏向性κ激动剂可以减轻疼痛和瘙痒,但副作用较少,如快感缺失和精神病,而这些副作用迄今为止限制了非偏向性κ阿片激动剂的临床开发。在此,我们总结最近发现的G蛋白偏向性κ激动剂,比较它们的结构、信号偏向程度和临床前效应。我们特别回顾了纳呋拉啡、22-硫氰酸萨维诺林A(RB-64)、甲磺酰基萨维诺林B、2-(4-(呋喃-2-基甲基)-5-((4-甲基-3-(三氟甲基)苄基)硫代)-4H-1,2,4-三唑-3-基)吡啶(三唑1.1)、3-(2-((环丙基甲基)(苯乙基)氨基)乙基)苯酚(HS666)、叔丁基-苯乙基-3-羟基苯乙胺(化合物5/BPHA)、6-胍基纳曲酮(6'GNTI)和collybolide。这些激动剂包含多种化学骨架,在G蛋白信号传导和β-抑制蛋白招募方面的效力和功效各不相同。因此,毫不奇怪,这些激动剂报告的行为反应并不一致。然而,我们的结论是,κ阿片领域将从未来比较几种偏向性激动剂并将信号偏向程度与特定药理反应相关联的研究中受益匪浅。