Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), 1 rue Laurent Fries, 67404, Illkirch, France.
INSERM U1258, Illkirch, France.
Acta Neuropathol. 2019 Oct;138(4):631-652. doi: 10.1007/s00401-019-02017-9. Epub 2019 May 7.
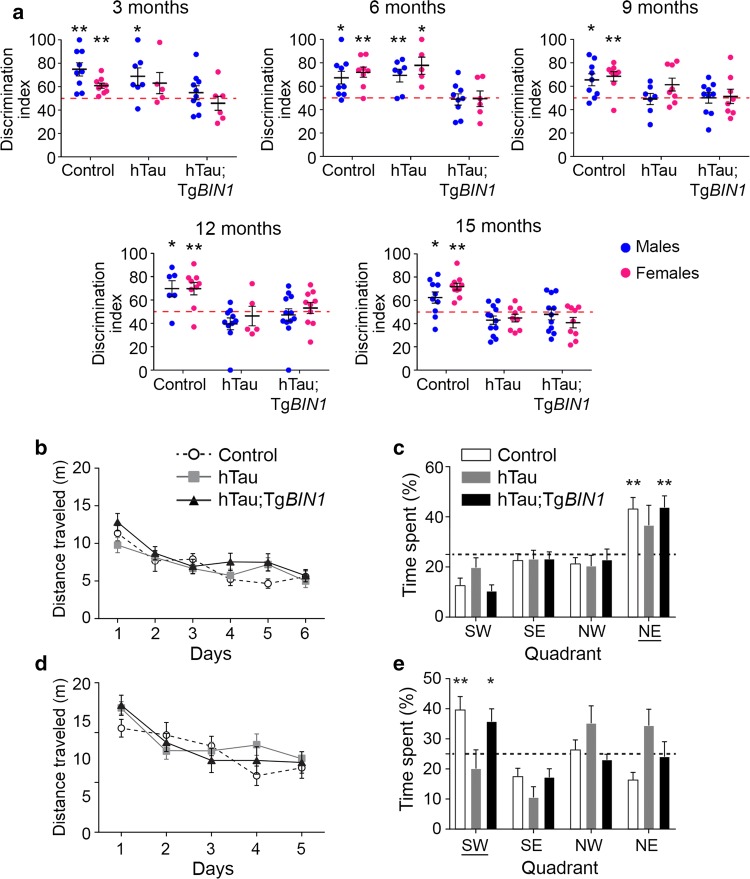
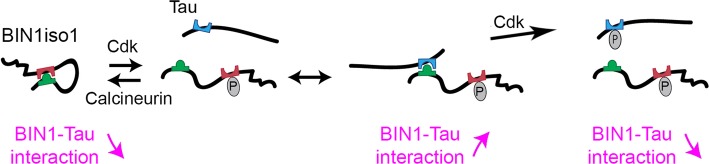
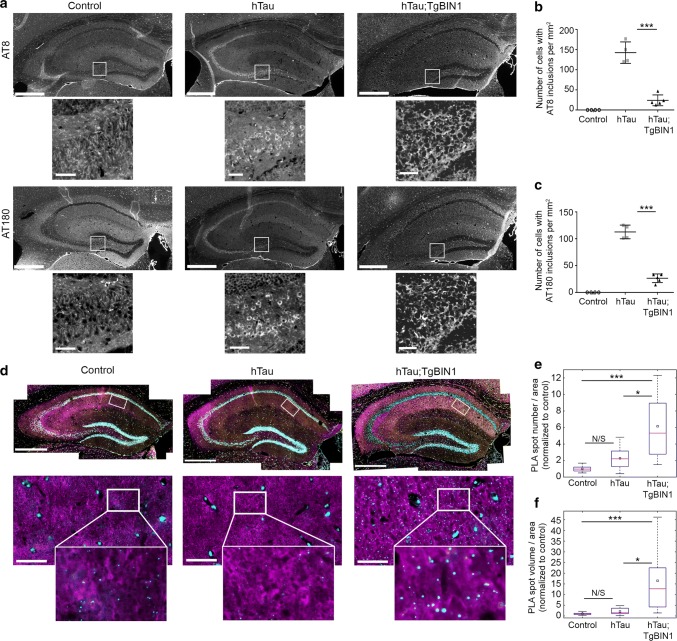
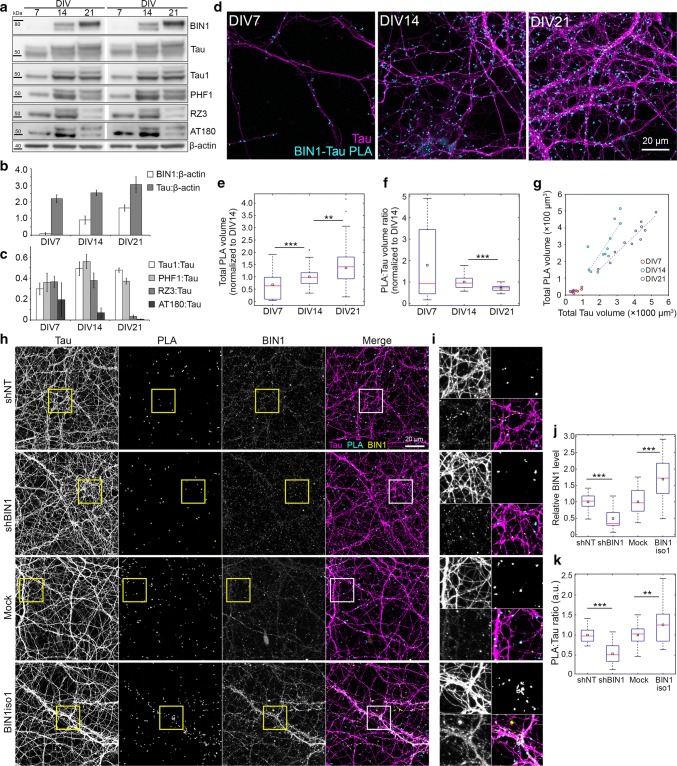
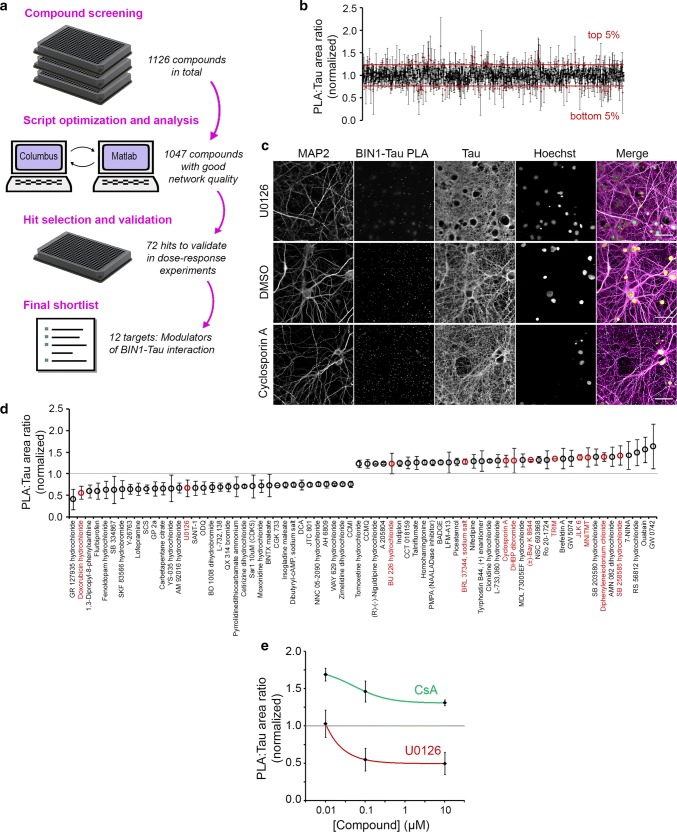
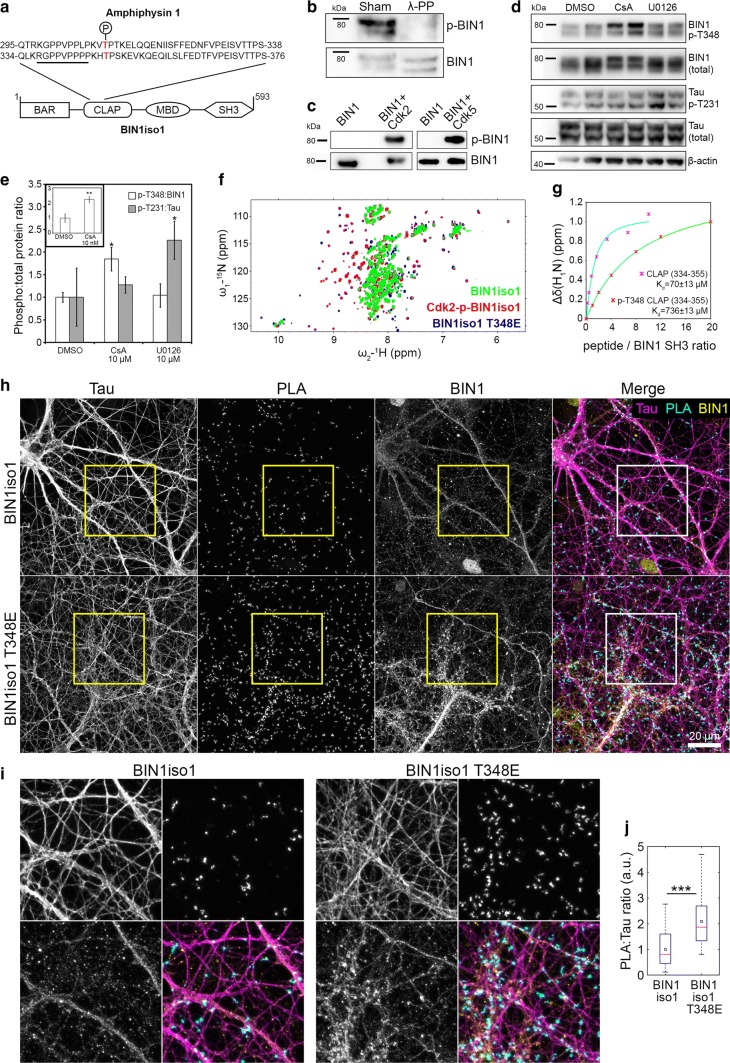
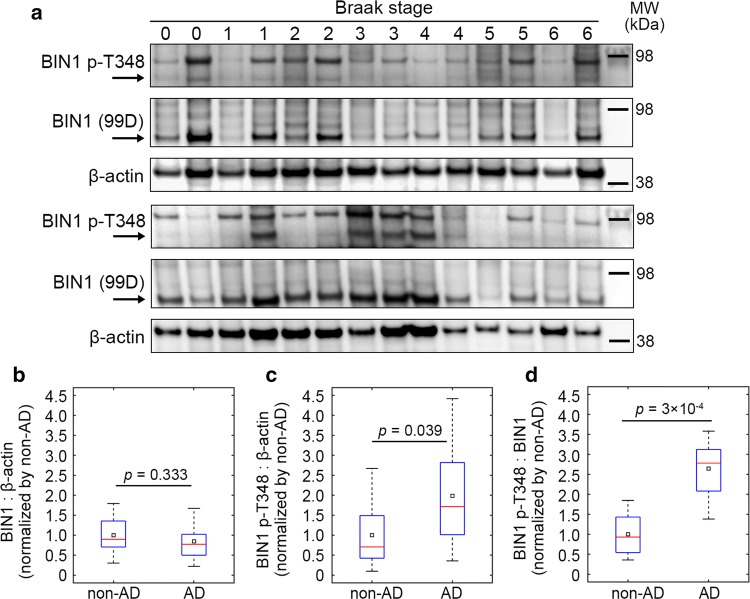
The bridging integrator 1 gene (BIN1) is a major genetic risk factor for Alzheimer's disease (AD). In this report, we investigated how BIN1-dependent pathophysiological processes might be associated with Tau. We first generated a cohort of control and transgenic mice either overexpressing human MAPT (TgMAPT) or both human MAPT and BIN1 (TgMAPT;TgBIN1), which we followed-up from 3 to 15 months. In TgMAPT;TgBIN1 mice short-term memory deficits appeared earlier than in TgMAPT mice; however-unlike TgMAPT mice-TgMAPT;TgBIN1 mice did not exhibit any long-term or spatial memory deficits for at least 15 months. After killing the cohort at 18 months, immunohistochemistry revealed that BIN1 overexpression prevents both Tau mislocalization and somatic inclusion in the hippocampus, where an increase in BIN1-Tau interaction was also observed. We then sought mechanisms controlling the BIN1-Tau interaction. We developed a high-content screening approach to characterize modulators of the BIN1-Tau interaction in an agnostic way (1,126 compounds targeting multiple pathways), and we identified-among others-an inhibitor of calcineurin, a Ser/Thr phosphatase. We determined that calcineurin dephosphorylates BIN1 on a cyclin-dependent kinase phosphorylation site at T348, promoting the open conformation of the neuronal BIN1 isoform. Phosphorylation of this site increases the availability of the BIN1 SH3 domain for Tau interaction, as demonstrated by nuclear magnetic resonance experiments and in primary neurons. Finally, we observed that although the levels of the neuronal BIN1 isoform were unchanged in AD brains, phospho-BIN1(T348):BIN1 ratio was increased, suggesting a compensatory mechanism. In conclusion, our data support the idea that BIN1 modulates the AD risk through an intricate regulation of its interaction with Tau. Alteration in BIN1 expression or activity may disrupt this regulatory balance with Tau and have direct effects on learning and memory.
桥接整合因子 1 基因(BIN1)是阿尔茨海默病(AD)的主要遗传风险因素。在本报告中,我们研究了 BIN1 依赖性病理生理过程如何与 Tau 相关。我们首先生成了一组对照和转基因小鼠队列,这些小鼠要么过表达人 MAPT(TgMAPT),要么同时过表达人 MAPT 和 BIN1(TgMAPT;TgBIN1),我们从 3 个月到 15 个月对其进行了跟踪。在 TgMAPT;TgBIN1 小鼠中,短期记忆缺陷出现得比 TgMAPT 小鼠更早;然而-与 TgMAPT 小鼠不同-TgMAPT;TgBIN1 小鼠在至少 15 个月内没有表现出任何长期或空间记忆缺陷。在 18 个月时杀死该队列后,免疫组织化学显示 BIN1 过表达可防止 Tau 定位错误和体部包含在内,在海马体中还观察到 BIN1-Tau 相互作用增加。然后,我们寻找控制 BIN1-Tau 相互作用的机制。我们开发了一种高通量筛选方法,以一种无偏见的方式(针对多种途径的 1,126 种化合物)来表征 BIN1-Tau 相互作用的调节剂,我们确定了-除其他外-钙调神经磷酸酶抑制剂。我们确定钙调神经磷酸酶在 T348 处对 BIN1 进行环依赖性激酶磷酸化,从而促进神经元 BIN1 同工型的开放构象。通过核磁共振实验和原代神经元证实,该位点的磷酸化增加了 BIN1 SH3 结构域与 Tau 相互作用的可用性。最后,我们观察到,尽管 AD 大脑中的神经元 BIN1 同工型水平没有改变,但磷酸化 BIN1(T348):BIN1 比值增加,表明存在代偿机制。总之,我们的数据支持 BIN1 通过其与 Tau 的相互作用的复杂调节来调节 AD 风险的观点。BIN1 表达或活性的改变可能会破坏与 Tau 的这种调节平衡,并对学习和记忆产生直接影响。