School of Science, Jiangnan University, Wuxi, 214122, China.
Wuxi Engineering Research Center for Biocomputing, Wuxi, 214122, China.
BMC Bioinformatics. 2019 May 1;20(Suppl 7):197. doi: 10.1186/s12859-019-2739-z.
Lung adenocarcinoma is the most common type of lung cancer, with high mortality worldwide. Its occurrence and development were thoroughly studied by high-throughput expression microarray, which produced abundant data on gene expression, DNA methylation, and miRNA quantification. However, the hub genes, which can be served as bio-markers for discriminating cancer and healthy individuals, are not well screened.
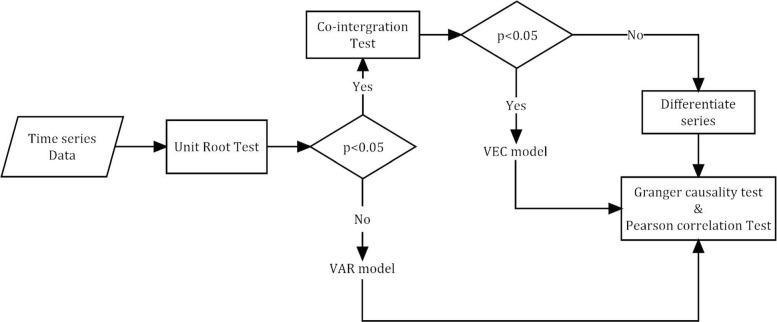
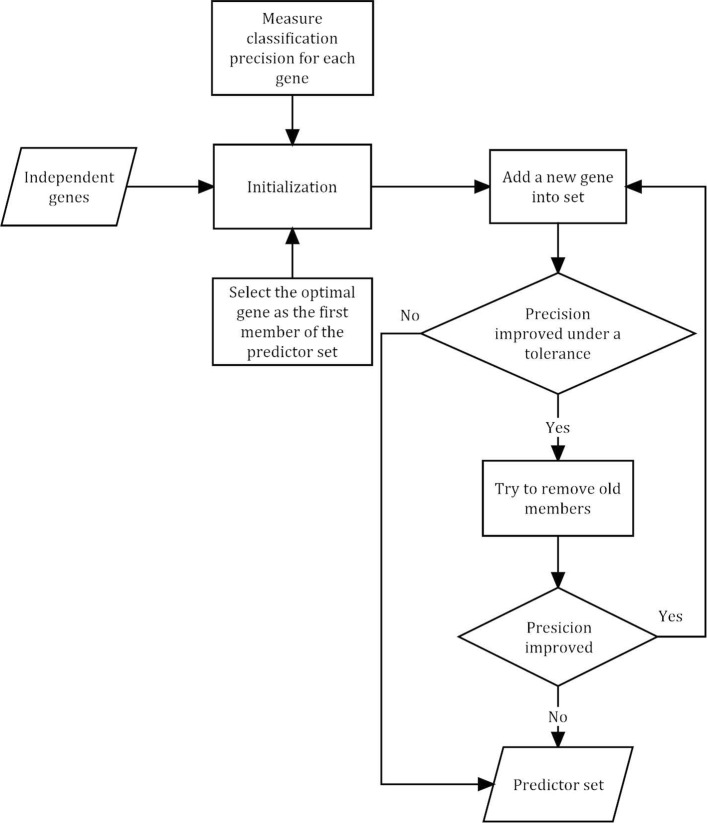
Here we present a new method for extracting gene predictors, aiming to obtain the least predictors without losing the efficiency. We firstly analyzed three different expression microarrays and constructed multi-interaction network, since the individual expression dataset is not enough for describing biological behaviors dynamically and systematically. Then, we transformed the undirected interaction network to directed network by employing Granger causality test, followed by the predictors screened with the use of the stepwise character selection algorithm. Six predictors, including TOP2A, GRK5, SIRT7, MCM7, EGFR, and COL1A2, were ultimately identified. All the predictors are the cancer-related, and the number is very small fascinating diagnosis. Finally, the validation of this approach was verified by robustness analyses applied to six independent datasets; the precision is up to 95.3% ∼ 100%.
Although there are complicated differences between cancer and normal cells in gene functions, cancer cells could be differentiated in case that a group of special genes expresses abnormally. Here we presented a new, robust, and effective method for extracting gene predictors. We identified as low as 6 genes which can be taken as predictors for diagnosing lung adenocarcinoma.
肺腺癌是最常见的肺癌类型,全球死亡率很高。高通量表达微阵列对其发生发展进行了深入研究,产生了大量关于基因表达、DNA 甲基化和 miRNA 定量的数据。然而,作为区分癌症和健康个体的生物标志物的枢纽基因尚未得到很好的筛选。
我们在这里提出了一种新的提取基因预测因子的方法,旨在获得最少的预测因子而不降低效率。我们首先分析了三个不同的表达微阵列,并构建了多相互作用网络,因为个体表达数据集不足以动态和系统地描述生物行为。然后,我们通过使用格兰杰因果关系检验将无向相互作用网络转换为有向网络,然后使用逐步特征选择算法筛选预测因子。最终确定了六个预测因子,包括 TOP2A、GRK5、SIRT7、MCM7、EGFR 和 COL1A2。所有的预测因子都是与癌症相关的,而且数量非常少,令人着迷的诊断。最后,通过应用稳健性分析对六个独立数据集进行了该方法的验证;精度高达 95.3%∼100%。
尽管癌细胞在基因功能上存在复杂的差异,但如果一组特殊基因异常表达,就可以将其区分出来。在这里,我们提出了一种新的、稳健的、有效的提取基因预测因子的方法。我们确定了低至 6 个基因可以作为肺腺癌诊断的预测因子。