Yang Mengxue, Sun Bowen, Li Jianhui, Yang Bo, Xu Jie, Zhou Xue, Yu Jie, Zhang Xuan, Zhang Qun, Zhou Shan, Sun Xiaohua
Department of Endocrinology, The Fifth People's Hospital of Shanghai, Fudan University, Shanghai, China.
Department of Endocrinology, Zunyi Medical University, Zunyi, China.
Endocr Connect. 2019 Jul;8(7):822-828. doi: 10.1530/EC-19-0001.
The pathogenesis of Graves' disease (GD) remains unclear. In terms of environmental factors, GD development may be associated with chronic inflammation caused by alteration of the intestinal flora. This study explored the association of intestinal flora alteration with the development of GD among the Han population in southwest China.
Fifteen GD patients at the Affiliated Hospital of Zunyi Medical College between March 2016 and March 2017 were randomly enrolled. Additionally, 15 sex- and age-matched healthy volunteers were selected as the control group during the same period. Fresh stool samples were collected, and bacterial 16S RNA was extracted and amplified for gene sequencing with the Illumina MiSeq platform. The sequencing results were subjected to operational taxonomic unit-based classification, classification verification, alpha diversity analysis, taxonomic composition analysis and partial least squares-discriminant analysis (PLS-DA).
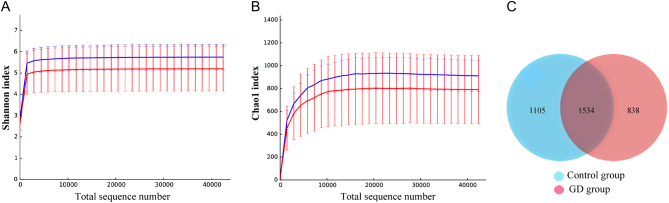
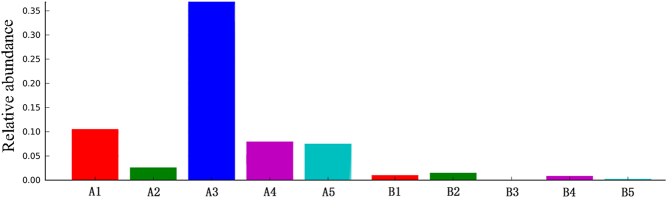
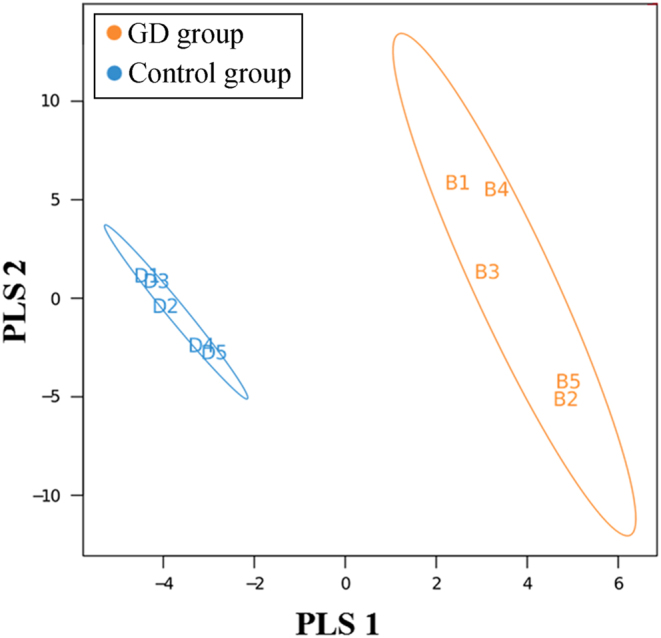
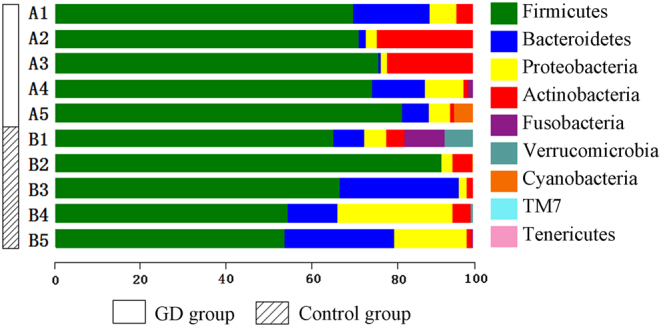
The diversity indices for the GD group were lower than those for the control group. The GD group showed significantly higher abundances of Firmicutes, Proteobacteria and Actinobacillus and a higher Firmicutes/Bacteroidetes ratio than the control group. PLS-DA suggested the satisfactory classification of the flora between the GD group and the control group. The abundances of the genera Oribacterium, Mogibacterium, Lactobacillus, Aggregatibacter and Mogibacterium were significantly higher in the GD group than in the control group (P < 0.05).
The intestinal flora of GD patients was significantly different from that of the healthy population. Thus, alteration of intestinal flora may be associated with the development of GD.
Graves病(GD)的发病机制尚不清楚。就环境因素而言,GD的发生可能与肠道菌群改变引起的慢性炎症有关。本研究探讨了中国西南地区汉族人群肠道菌群改变与GD发生之间的关联。
随机纳入2016年3月至2017年3月期间遵义医学院附属医院的15例GD患者。此外,同期选取15名年龄和性别匹配的健康志愿者作为对照组。收集新鲜粪便样本,提取细菌16S RNA并进行扩增,以使用Illumina MiSeq平台进行基因测序。对测序结果进行基于操作分类单元的分类、分类验证、α多样性分析、分类组成分析和偏最小二乘判别分析(PLS-DA)。
GD组的多样性指数低于对照组。GD组的厚壁菌门、变形菌门和放线杆菌属的丰度显著高于对照组,且厚壁菌门/拟杆菌门的比例也高于对照组。PLS-DA表明GD组和对照组之间的菌群分类良好。GD组中Oribacterium、Mogibacterium、乳杆菌属、聚集杆菌属和Mogibacterium属的丰度显著高于对照组(P < 0.05)。
GD患者的肠道菌群与健康人群有显著差异。因此,肠道菌群改变可能与GD的发生有关。