Kioroglou Dimitrios, Mas Albert, Portillo Maria Del Carmen
Department Biochemistry and Biotechnology, Faculty of Oenology, University Rovira i Virgili, Tarragona, Spain.
Front Microbiol. 2019 May 16;10:1084. doi: 10.3389/fmicb.2019.01084. eCollection 2019.
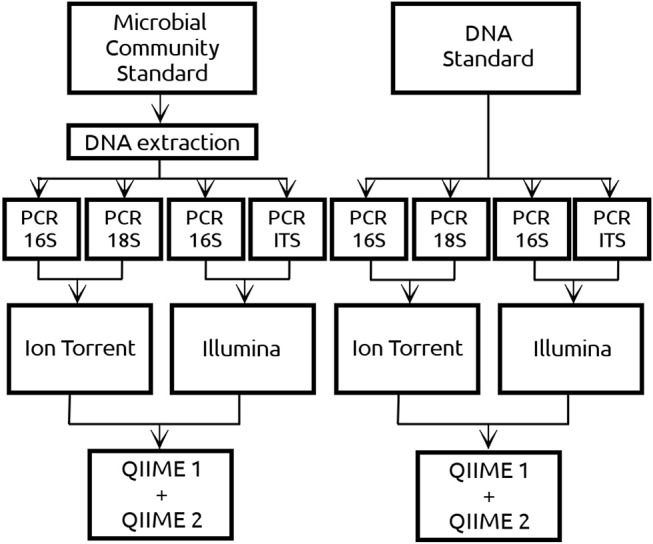
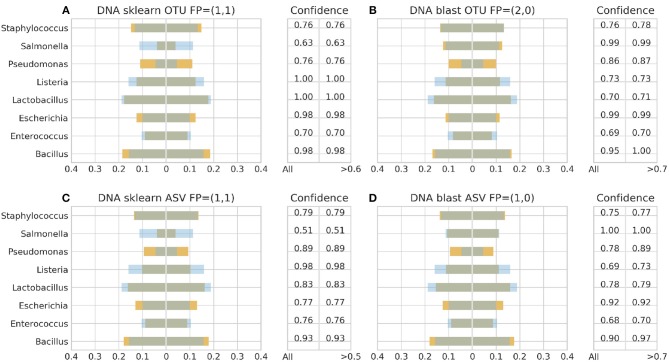
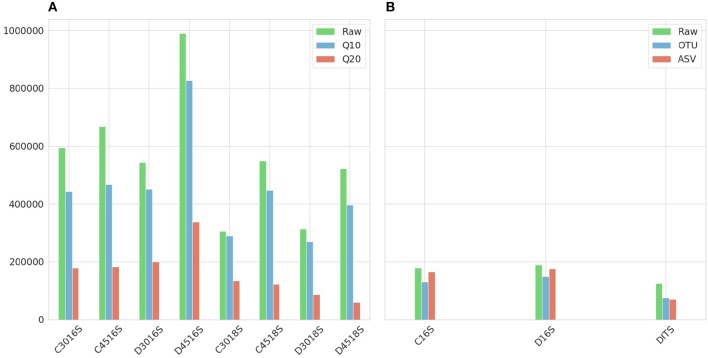
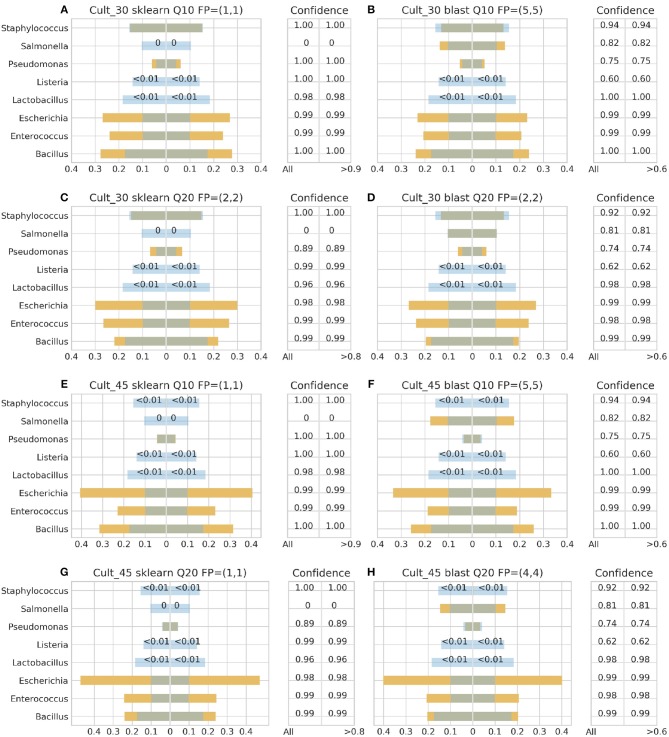
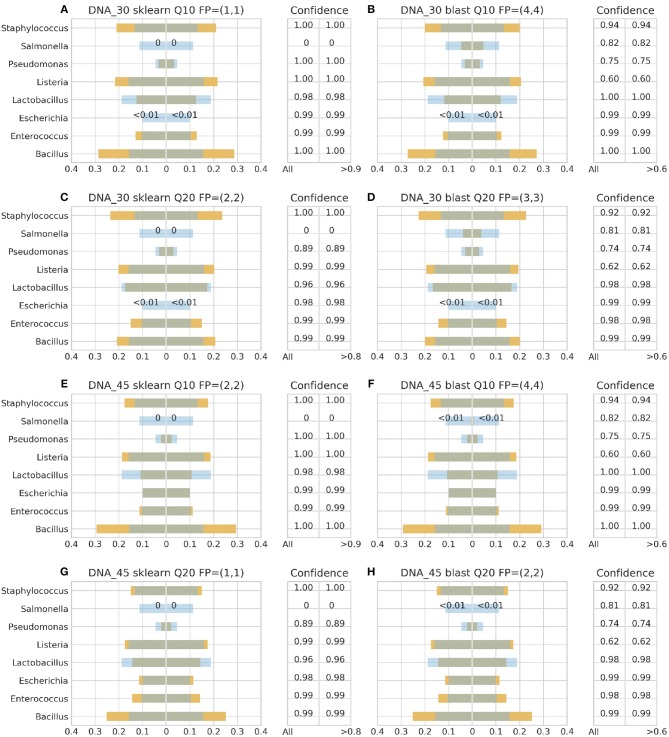
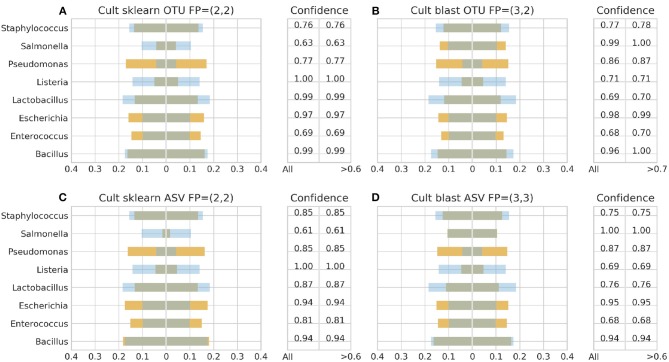
Metataxonomic analysis represents a fast and cost-effective approach for acquiring informative insight into the composition of the microbiome of samples with variable diversity, such as wine samples. Nevertheless, it comprises a vast amount of laboratory procedures and bioinformatic frameworks each one associated with an inherent variability of protocols and algorithms, respectively. As a solution to the bioinformatic maze, QIIME bioinformatic framework has incorporated benchmarked, and balanced parameters as default parameters. In the current study, metataxonomic analysis of two types of mock community standards with the same microbial composition has been performed for evaluating the effectivess of QIIME balanced default parameters on a variety of aspects related to different laboratory and bioinformatic workflows. These aspects concern NGS platforms, PCR protocols, bioinformatic pipelines, and taxonomic classification algorithms. Several qualitative performance expectations have been the outcome of the analysis, rendering the mock community a useful evaluation tool.
宏分类学分析是一种快速且经济高效的方法,可用于深入了解具有不同多样性的样本(如葡萄酒样本)的微生物群落组成。然而,它包含大量实验室程序和生物信息学框架,每个程序和框架分别与协议和算法的固有变异性相关。作为解决生物信息学迷宫的方案,QIIME生物信息学框架已纳入经过基准测试且平衡的参数作为默认参数。在本研究中,对具有相同微生物组成的两种类型的模拟群落标准进行了宏分类学分析,以评估QIIME平衡默认参数在与不同实验室和生物信息学工作流程相关的各个方面的有效性。这些方面涉及二代测序平台、PCR协议、生物信息学管道和分类算法。分析得出了一些定性的性能预期,使模拟群落成为一种有用的评估工具。