Department of Pharmaceutical Sciences, College of Clinical Pharmacy, King Faisal University, Al-Ahsa, Kingdom of Saudi Arabia.
Department of Biotechnology and Food Technology, Durban University of Technology, Durban, South Africa.
PLoS One. 2019 Jun 4;14(6):e0217270. doi: 10.1371/journal.pone.0217270. eCollection 2019.
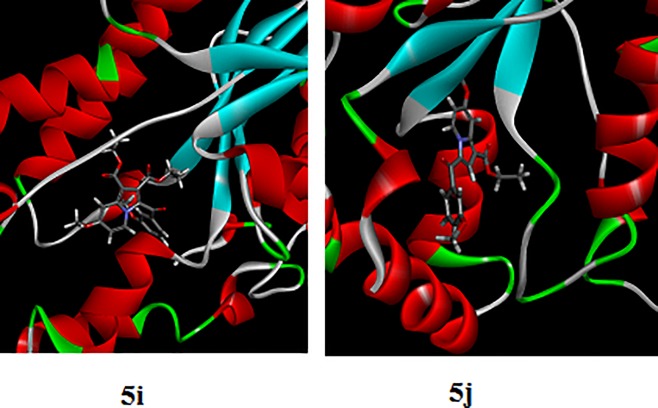
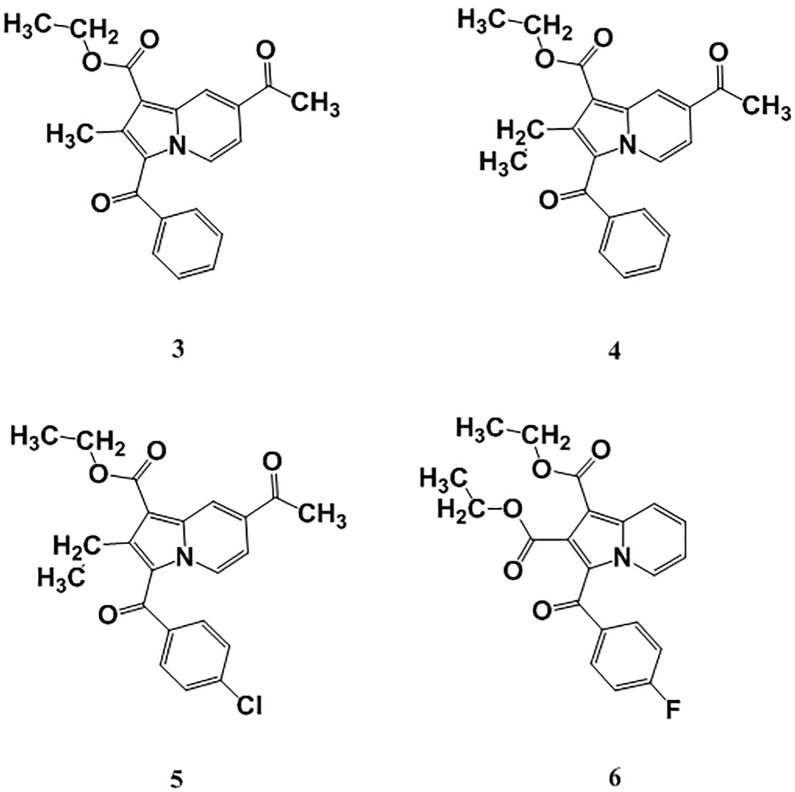
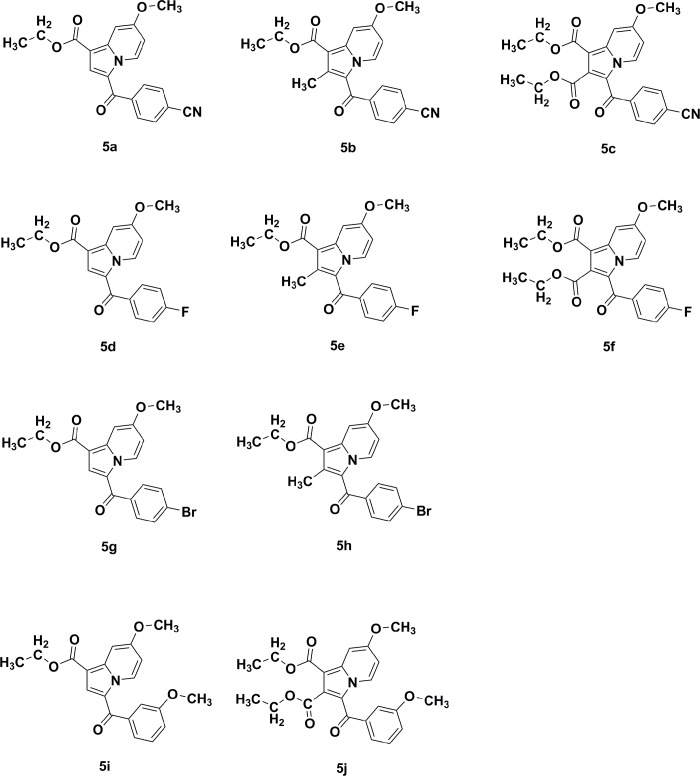
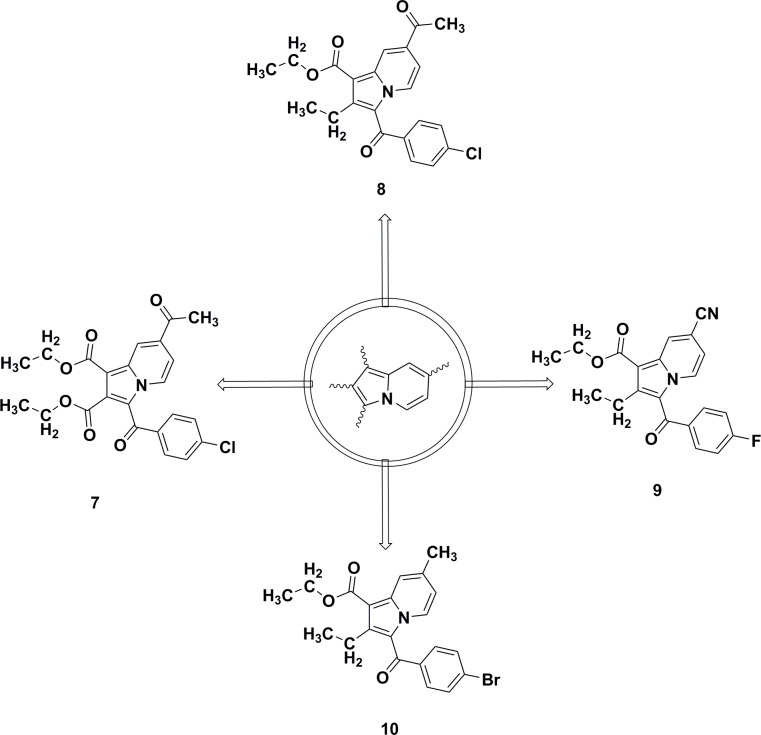
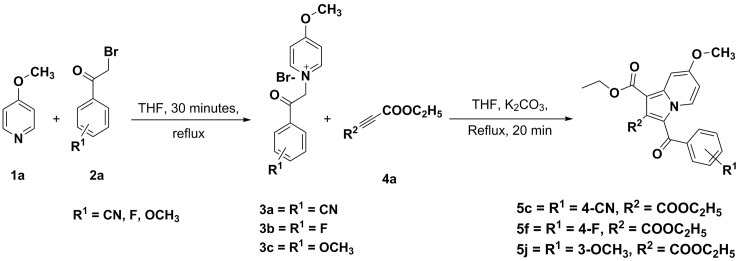
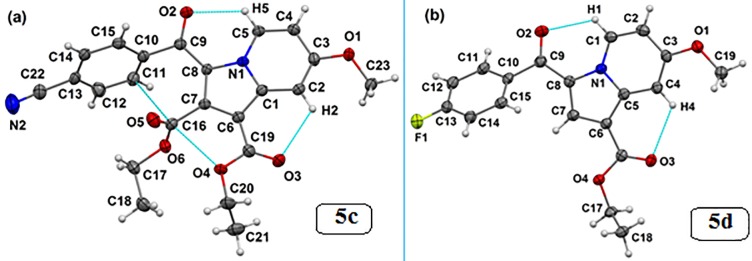
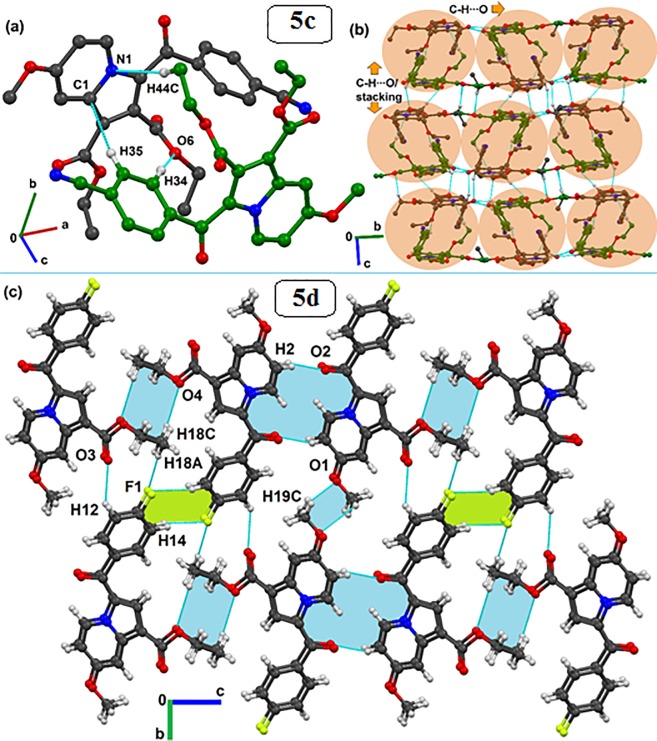
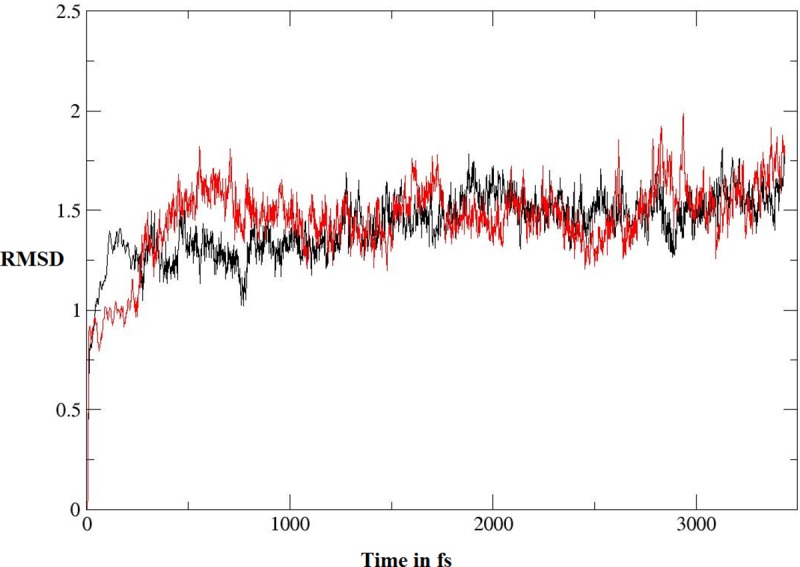
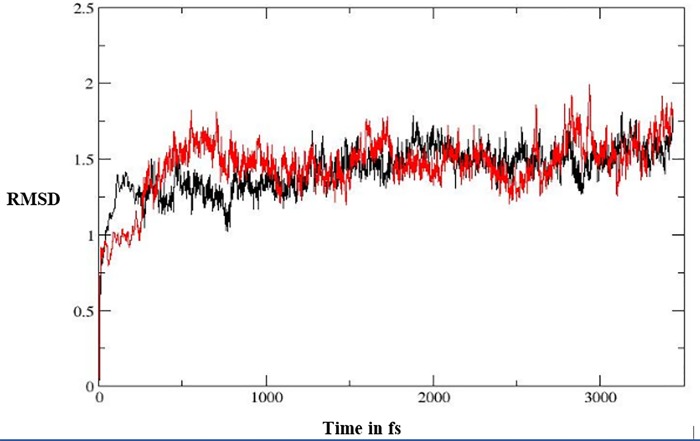
Indolizines are heteroaromatic compounds, and their synthetic analogues have reportedly showed promising pharmacological properties. In this study, a series of synthetic 7-methoxy-indolizine derivatives were synthesised, characterised and evaluated for in vitro whole-cell anti-tuberculosis (TB) screening against susceptible (H37Rv) and multi-drug-resistant (MDR) strains of Mycobacterium tuberculosis (MTB) using the resazurin microplate assay method. The cytotoxicity was evaluated using the MTT assay. In silico molecular-docking study was conducted for compounds 5a-j against enoyl-[acyl-carrier] protein reductase, a key enzyme of the type II fatty acid synthesis that has attracted much interest for the development of novel anti-TB compounds. Thereafter, molecular dynamic (MD) simulation was undertaken for the most active inhibitors. Compounds 5i and 5j with the methoxy functional group at the meta position of the benzoyl group, which was at the third position of the indolizine nucleus, demonstrated encouraging anti-TB activity against MDR strains of MTB at 16 μg/mL. In silico studies showed binding affinity within the range of 7.07-8.57 kcal/mol, with 5i showing the highest binding affinity. Hydrogen bonding, π-π- interactions, and electrostatic interactions were common with the active site. Most of these interactions occurred with the catalytic amino acids (Pro193, Tyr158, Phe149, and Lys165). MD simulation showed that 5j possessed the highest binding affinity toward the enzyme, according to the two calculation methods (MM/PBSA and MM/GBSA). The single-crystal X-ray studies of compounds 5c and 5d revealed that the molecular arrangements in these two structures were mostly guided by C-H···O hydrogen-bonded dimeric motifs and C-H···N hydrogen bonds, while various secondary interactions (such as π···π and C-H···F) also contributed to crystal formation. Compounds 5a, 5c, 5i, and 5j exhibited no toxicity up to 500 μg/mL. In conclusion, 5i and 5j are promising anti-TB compounds that have shown high affinity based on docking and MD simulation results.
吲哚嗪是杂芳族化合物,其合成类似物据称表现出有前景的药理学特性。在这项研究中,合成了一系列合成的 7-甲氧基吲哚嗪衍生物,并通过使用 Resazurin 微量板测定法对体外全细胞抗结核(TB)筛选进行了评估,针对敏感(H37Rv)和多药耐药(MDR)结核分枝杆菌(MTB)菌株。使用 MTT 测定法评估细胞毒性。使用化合物 5a-j 对烯酰-[酰基载体]蛋白还原酶进行了计算机分子对接研究,烯酰-[酰基载体]蛋白还原酶是 II 型脂肪酸合成的关键酶,该酶因其对新型抗结核化合物的开发而备受关注。之后,对最活跃的抑制剂进行了分子动力学(MD)模拟。在吲哚嗪核的第三位置的苯甲酰基的间位具有甲氧基功能基团的化合物 5i 和 5j 对 16μg/mL 的 MDR 株 MTB 表现出令人鼓舞的抗结核活性。计算机研究表明,结合亲和力在 7.07-8.57 kcal/mol 范围内,其中 5i 表现出最高的结合亲和力。氢键、π-π-相互作用和静电相互作用与活性部位常见。大多数这些相互作用发生在催化氨基酸(Pro193、Tyr158、Phe149 和 Lys165)上。MD 模拟表明,根据两种计算方法(MM/PBSA 和 MM/GBSA),化合物 5j 对酶具有最高的结合亲和力。化合物 5c 和 5d 的单晶 X 射线研究表明,这两种结构中的分子排列主要受 C-H···O 氢键二聚体基序和 C-H···N 氢键的控制,而各种次级相互作用(如 π···π 和 C-H···F)也有助于晶体形成。化合物 5a、5c、5i 和 5j 在高达 500μg/mL 时没有表现出毒性。总之,5i 和 5j 是有前途的抗结核化合物,根据对接和 MD 模拟结果,它们表现出高亲和力。