Department of Biochemistry & Biophysics, Texas A&M University, College Station, 77843, TX, USA.
Molecular and Computational Biology Section, Department of Biological Sciences, University of Southern California, Los Angeles, 90089, CA, USA.
BMC Genomics. 2019 Jun 6;20(Suppl 5):425. doi: 10.1186/s12864-019-5702-5.
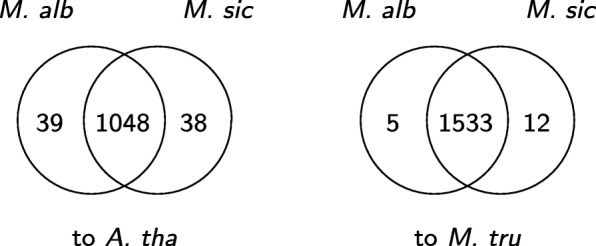
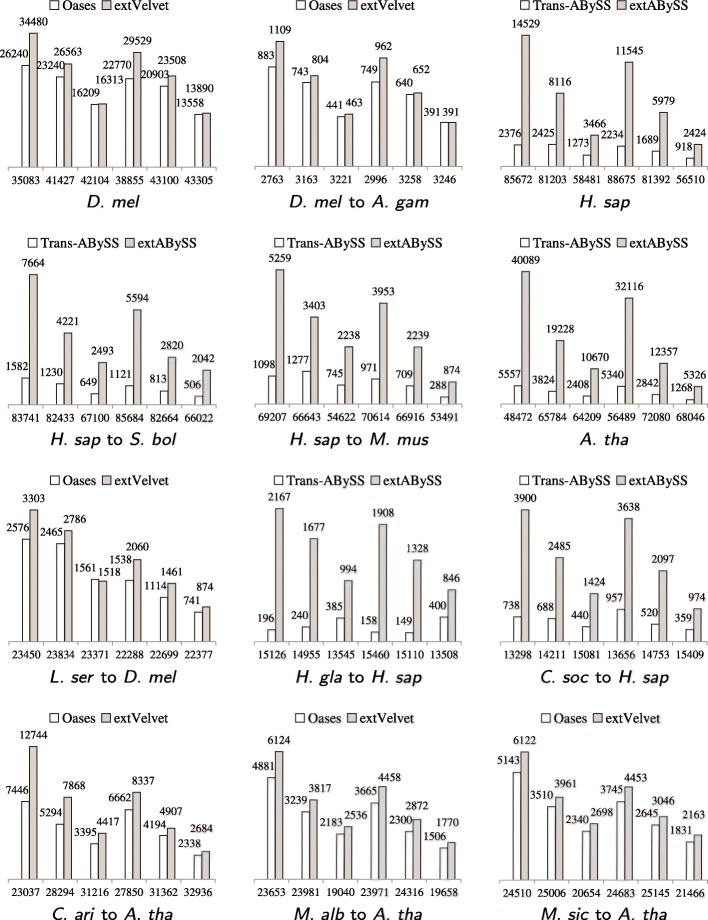
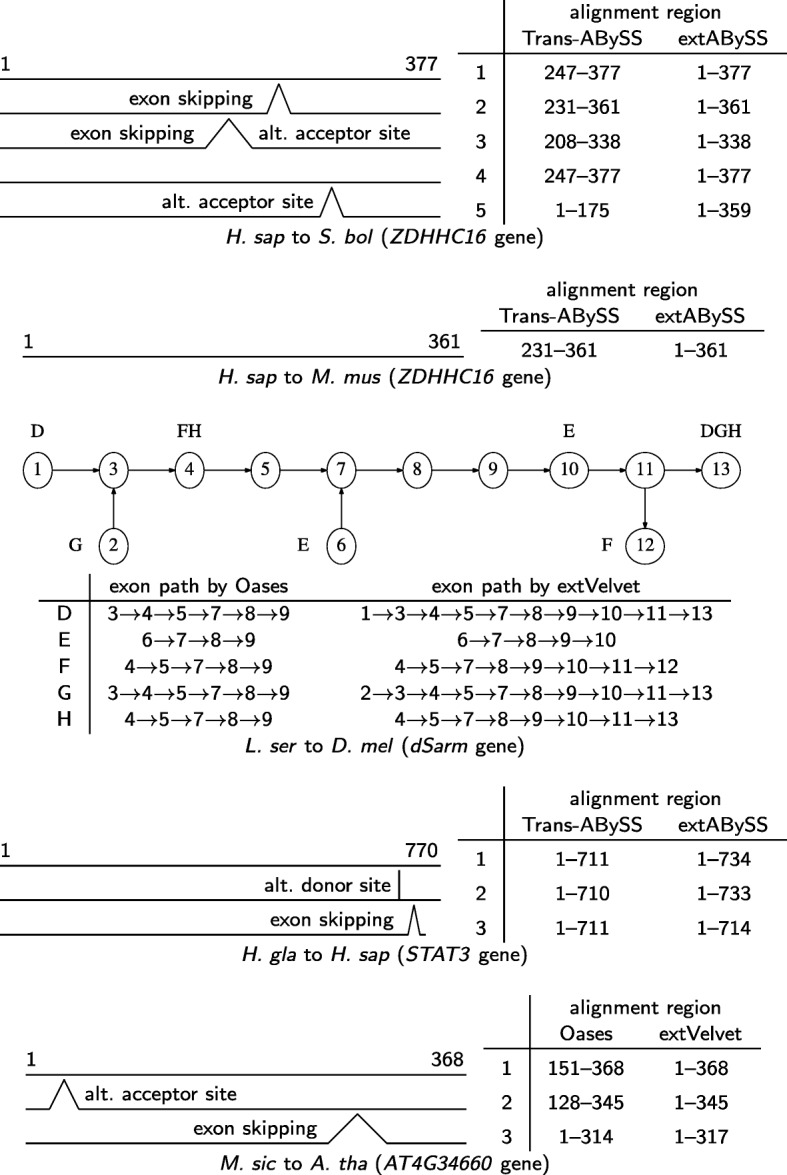
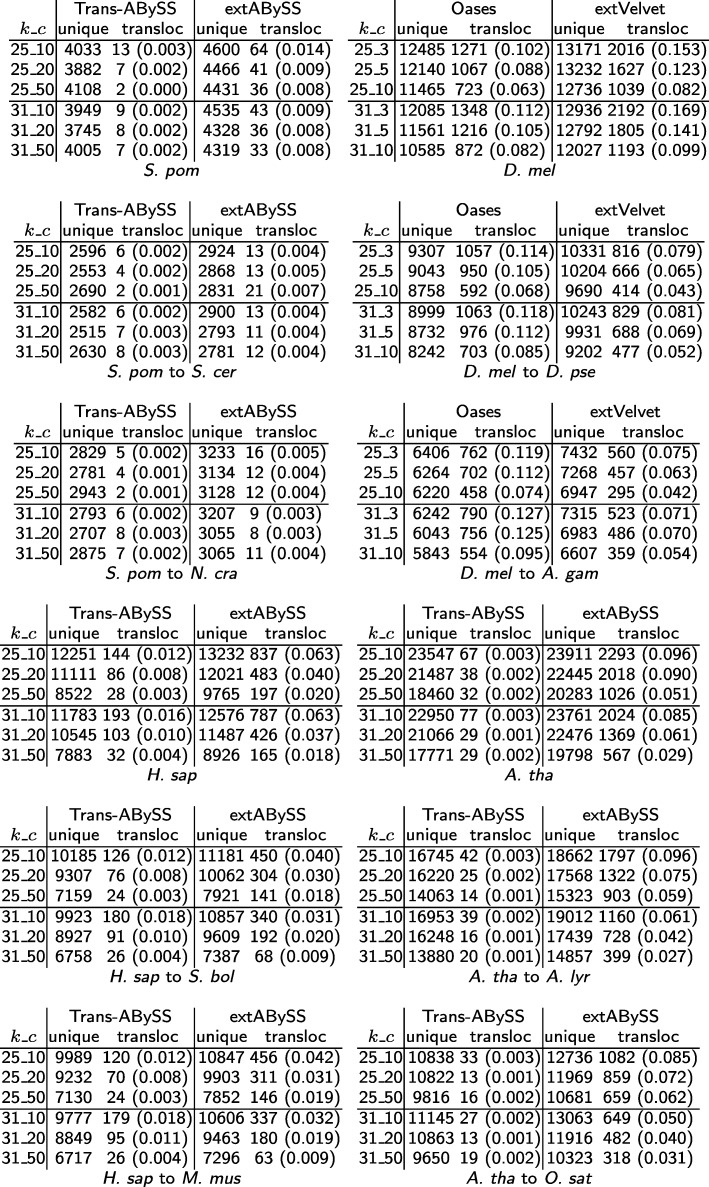
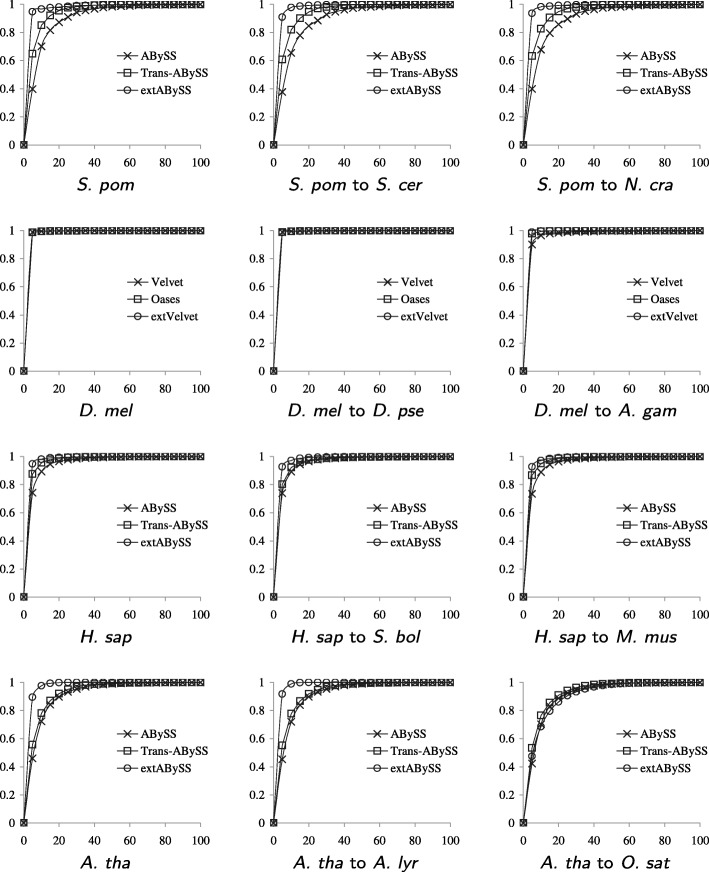
A popular strategy to study alternative splicing in non-model organisms starts from sequencing the entire transcriptome, then assembling the reads by using de novo transcriptome assembly algorithms to obtain predicted transcripts. A similarity search algorithm is then applied to a related organism to infer possible function of these predicted transcripts. While some of these predictions may be inaccurate and transcripts with low coverage are often missed, we observe that it is possible to obtain a more complete set of transcripts to facilitate possible functional assignments by starting the search from the intermediate de Bruijn graph that contains all branching possibilities.
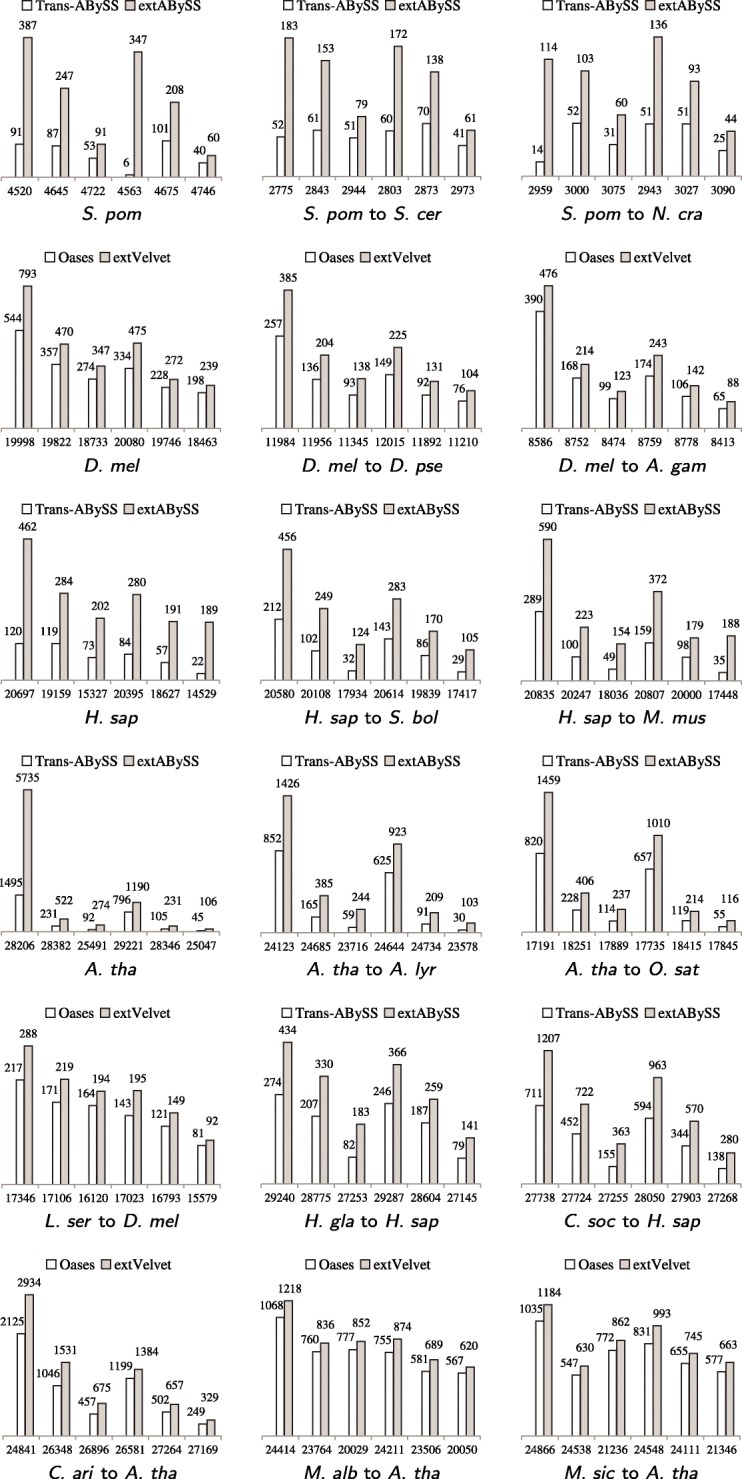
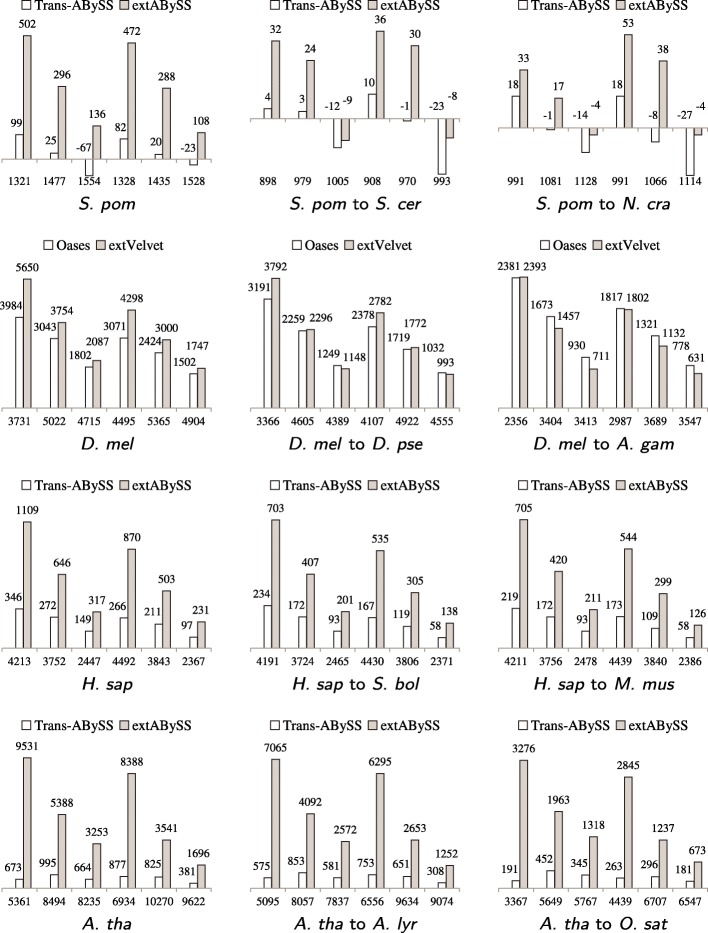
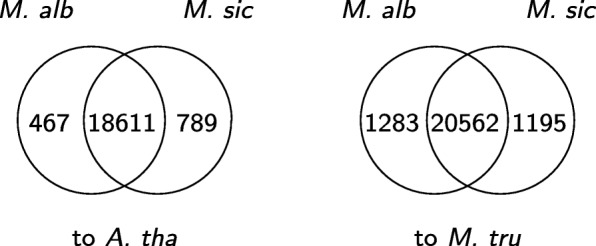
We develop an algorithm to extract similar transcripts in a related organism by starting the search from the de Bruijn graph that represents the transcriptome instead of from predicted transcripts. We show that our algorithm is able to recover more similar transcripts than existing algorithms, with large improvements in obtaining longer transcripts and a finer resolution of isoforms. We apply our algorithm to study salt and waterlogging tolerance in two Melilotus species by constructing new RNA-Seq libraries.
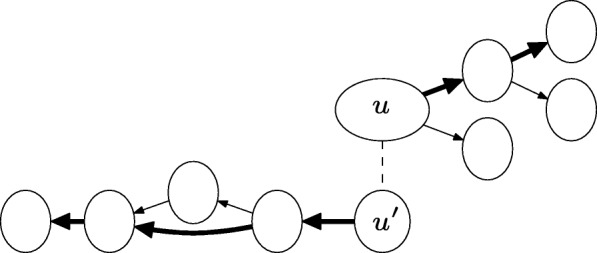
We have developed an algorithm to identify paths in the de Bruijn graph that correspond to similar transcripts in a related organism directly. Our strategy bypasses the transcript prediction step in RNA-Seq data and makes use of support from evolutionary information.
研究非模式生物中可变剪接的一种流行策略是从测序整个转录组开始,然后使用从头转录组组装算法来组装读取,以获得预测的转录本。然后应用相似性搜索算法到相关的生物体,以推断这些预测转录本的可能功能。虽然其中一些预测可能不准确,并且覆盖度低的转录本经常被忽略,但我们观察到,通过从包含所有分支可能性的中间 de Bruijn 图开始搜索,可以获得更完整的转录本集,从而有助于进行可能的功能分配。
我们开发了一种从代表转录组的 de Bruijn 图而不是从预测的转录本开始搜索,从而在相关生物体中提取相似转录本的算法。我们表明,我们的算法能够比现有算法恢复更多的相似转录本,在获得更长的转录本和更精细的同工型分辨率方面有很大的改进。我们通过构建新的 RNA-Seq 文库,应用我们的算法来研究两个草木樨属物种的耐盐和耐涝性。
我们已经开发了一种算法,可以直接在相关生物体的 de Bruijn 图中识别对应于相似转录本的路径。我们的策略绕过了 RNA-Seq 数据中的转录本预测步骤,并利用了进化信息的支持。