School of Informatics and Computing, Indiana University, Bloomington, IN 47405, USA.
Bioinformatics. 2012 Sep 15;28(18):i363-i369. doi: 10.1093/bioinformatics/bts388.
One of the difficulties in metagenomic assembly is that homologous genes from evolutionarily closely related species may behave like repeats and confuse assemblers. As a result, small contigs, each representing a short gene fragment, instead of complete genes, may be reported by an assembler. This further complicates annotation of metagenomic datasets, as annotation tools (such as gene predictors or similarity search tools) typically perform poorly on configs encoding short gene fragments.
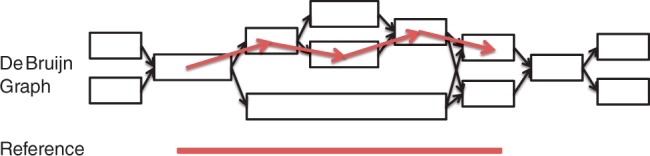
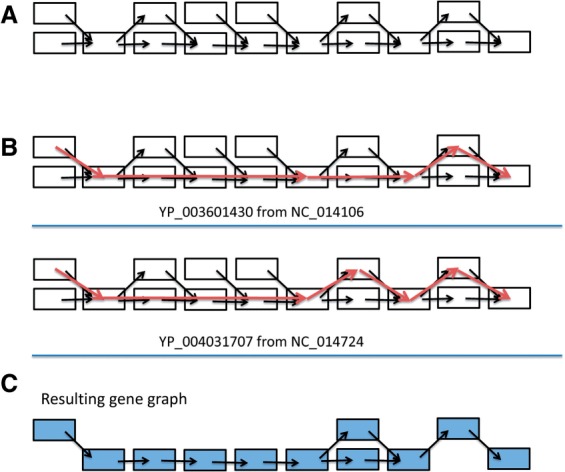
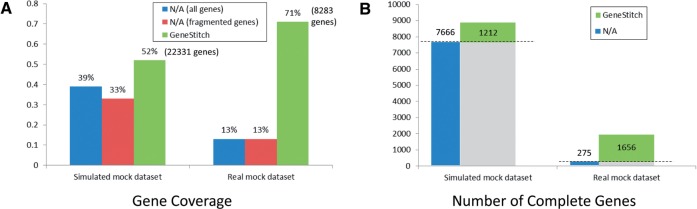
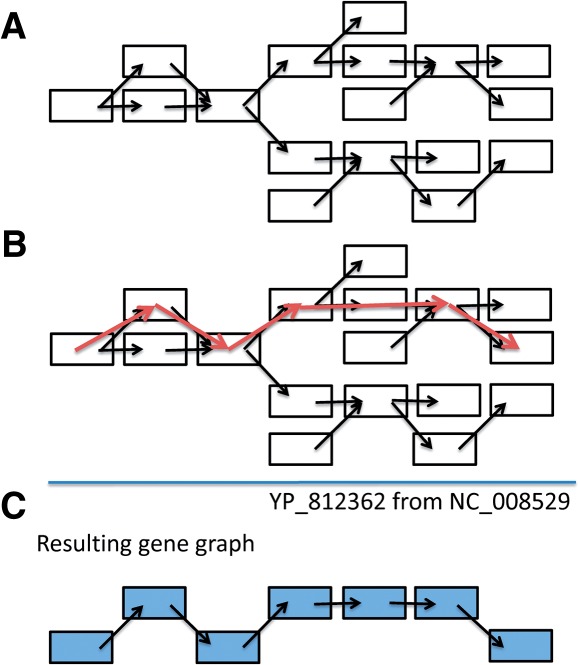
We present a novel way of using the de Bruijn graph assembly of metagenomes to improve the assembly of genes. A network matching algorithm is proposed for matching the de Bruijn graph of contigs against reference genes, to derive 'gene paths' in the graph (sequences of contigs containing gene fragments) that have the highest similarities to known genes, allowing gene fragments contained in multiple contigs to be connected to form more complete (or intact) genes. Tests on simulated and real datasets show that our approach (called GeneStitch) is able to significantly improve the assembly of genes from metagenomic sequences, by connecting contigs with the guidance of homologous genes-information that is orthogonal to the sequencing reads. We note that the improvement of gene assembly can be observed even when only distantly related genes are available as the reference. We further propose to use 'gene graphs' to represent the assembly of reads from homologous genes and discuss potential applications of gene graphs to improving functional annotation for metagenomics.
The tools are available as open source for download at http://omics.informatics.indiana.edu/GeneStitch
宏基因组组装的困难之一是,来自进化上密切相关的物种的同源基因可能表现得像重复序列,从而使组装器感到困惑。结果,组装器可能会报告小的 contigs,每个 contig 代表一个短的基因片段,而不是完整的基因。这进一步增加了宏基因组数据集注释的复杂性,因为注释工具(如基因预测器或相似性搜索工具)通常在对编码短基因片段的配置进行注释时表现不佳。
我们提出了一种利用宏基因组的 de Bruijn 图组装来改进基因组装的新方法。提出了一种网络匹配算法,用于将 contigs 的 de Bruijn 图与参考基因进行匹配,以从图中导出与已知基因具有最高相似性的“基因路径”(包含基因片段的 contig 序列),从而将包含在多个 contigs 中的基因片段连接起来,形成更完整(或完整)的基因。在模拟和真实数据集上的测试表明,我们的方法(称为 GeneStitch)能够通过使用同源基因的信息(与测序reads 正交的信息)来指导 contigs 的连接,从而显著改进宏基因组序列中基因的组装。我们注意到,即使只有远缘基因作为参考,也可以观察到基因组装的改进。我们进一步提出使用“基因图”来表示同源基因的 reads 组装,并讨论基因图在改进宏基因组功能注释方面的潜在应用。
该工具可作为开源软件从 http://omics.informatics.indiana.edu/GeneStitch 下载。