Department of Biomedical Informatics, The Ohio State University, Columbus, Ohio 43210, USA.
Department of Internal Medicine, University of Iowa, Iowa City, Iowa 52242, USA.
Genome Res. 2019 Aug;29(8):1329-1342. doi: 10.1101/gr.251116.119. Epub 2019 Jun 14.
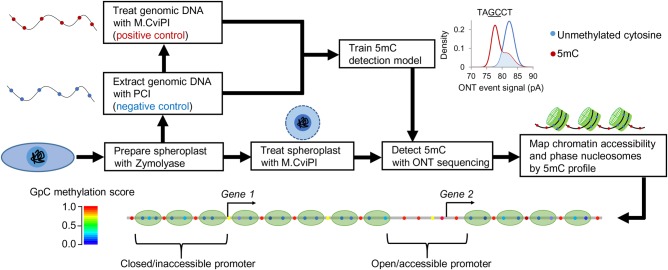
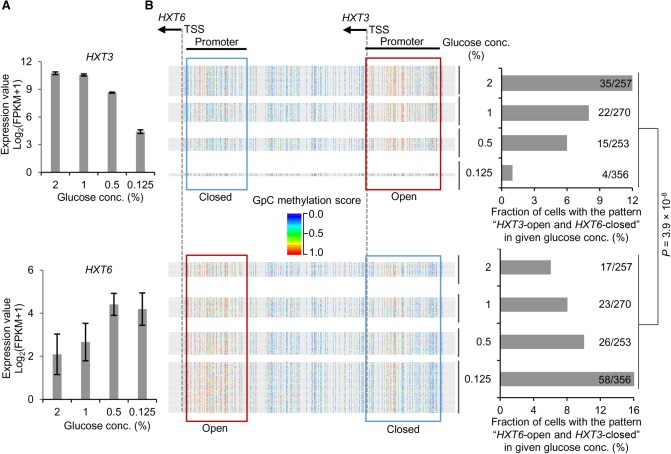
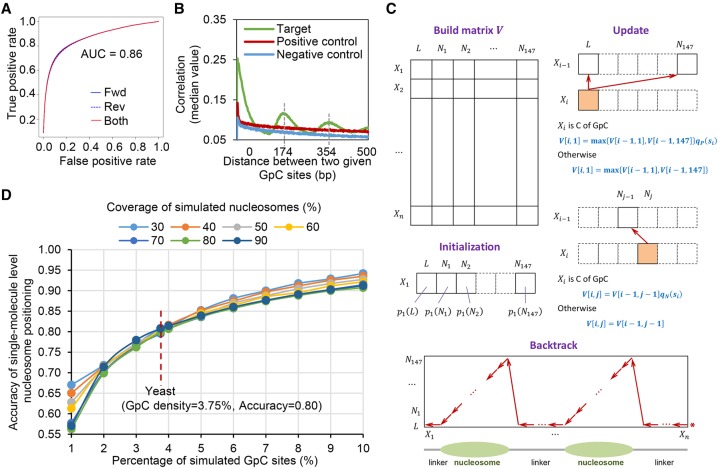
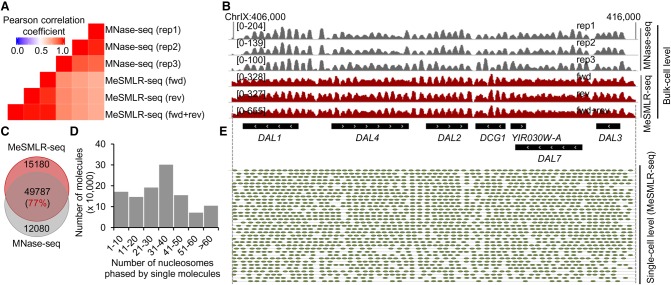
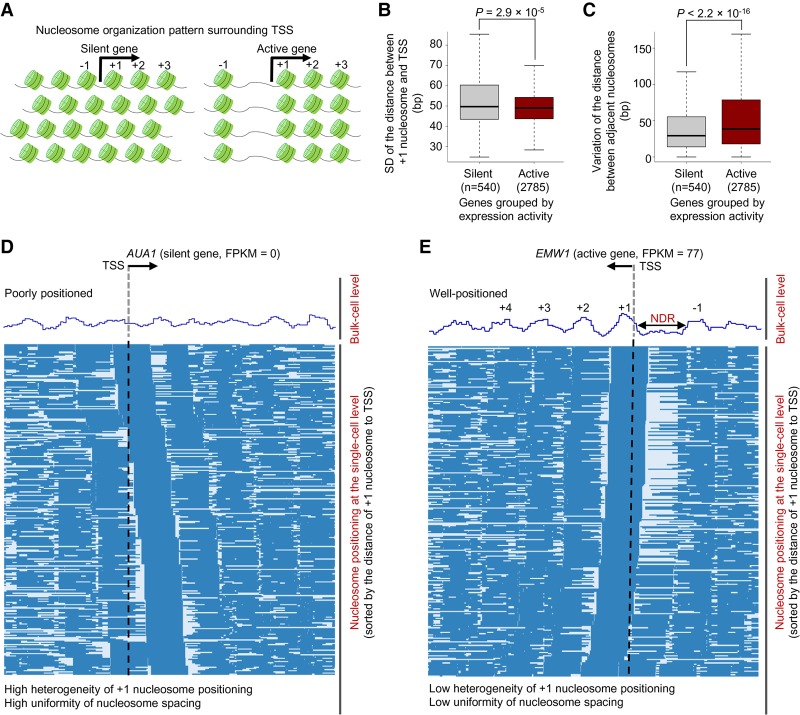
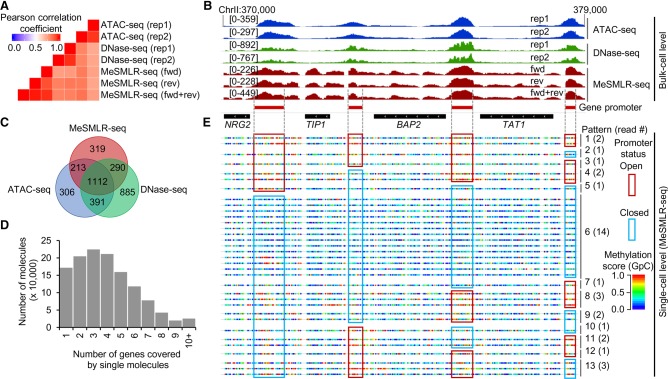
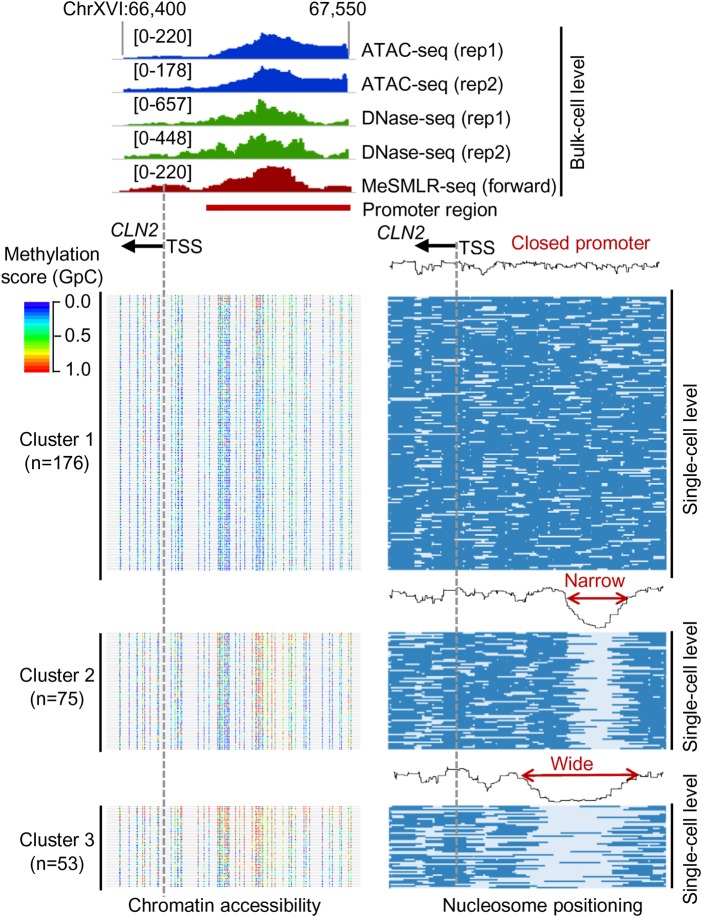
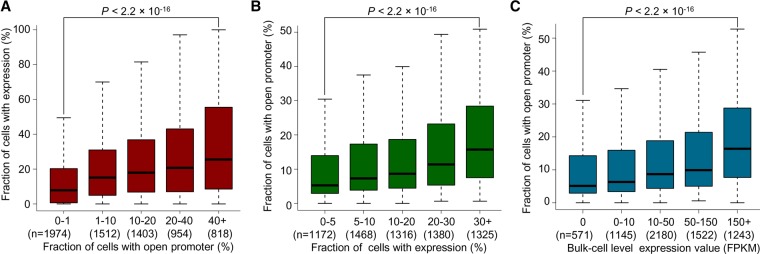
Genome-wide chromatin accessibility and nucleosome occupancy profiles have been widely investigated, while the long-range dynamics remain poorly studied at the single-cell level. Here, we present a new experimental approach, methyltransferase treatment followed by single-molecule long-read sequencing (MeSMLR-seq), for long-range mapping of nucleosomes and chromatin accessibility at single DNA molecules and thus achieve comprehensive-coverage characterization of the corresponding heterogeneity. MeSMLR-seq offers direct measurements of both nucleosome-occupied and nucleosome-evicted regions on a single DNA molecule, which is challenging for many existing methods. We applied MeSMLR-seq to haploid yeast, where single DNA molecules represent single cells, and thus we could investigate the combinatorics of many (up to 356) nucleosomes at long range in single cells. We illustrated the differential organization principles of nucleosomes surrounding the transcription start site for silent and actively transcribed genes, at the single-cell level and in the long-range scale. The heterogeneous patterns of chromatin status spanning multiple genes were phased. Together with single-cell RNA-seq data, we quantitatively revealed how chromatin accessibility correlated with gene transcription positively in a highly heterogeneous scenario. Moreover, we quantified the openness of promoters and investigated the coupled chromatin changes of adjacent genes at single DNA molecules during transcription reprogramming. In addition, we revealed the coupled changes of chromatin accessibility for two neighboring glucose transporter genes in response to changes in glucose concentration.
全基因组染色质可及性和核小体占有率图谱已经得到了广泛的研究,而单细胞水平的长程动力学仍研究甚少。在这里,我们提出了一种新的实验方法,即甲基转移酶处理后进行单分子长读测序(MeSMLR-seq),用于在单个 DNA 分子上对核小体和染色质可及性进行长程作图,从而实现对相应异质性的全面覆盖特征描述。MeSMLR-seq 提供了对单个 DNA 分子上核小体占据和核小体逐出区域的直接测量,这对于许多现有方法来说是具有挑战性的。我们将 MeSMLR-seq 应用于单倍体酵母中,其中单个 DNA 分子代表单个细胞,因此我们可以在单细胞中研究长程范围内多达 356 个核小体的组合。我们在单细胞水平和长程尺度上展示了转录起始位点周围沉默和活跃转录基因的核小体的不同组织原则。跨越多个基因的染色质状态的异质模式被分相。与单细胞 RNA-seq 数据相结合,我们定量地揭示了在高度异质的情况下,染色质可及性如何与基因转录呈正相关。此外,我们量化了启动子的开放性,并研究了转录重编程过程中单个 DNA 分子上相邻基因的偶联染色质变化。此外,我们揭示了两个相邻葡萄糖转运基因对葡萄糖浓度变化的染色质可及性的偶联变化。