Department of Anthropology, Faculty of Biology and Environmental Protection, University of Lodz, 12/16 Banacha Street, 90-237 Łódź, Poland.
Department of Molecular Biophysics, Faculty of Biology and Environmental Protection, University of Lodz, 12/16 Banacha Street, 90-237 Łódź, Poland.
Gigascience. 2019 Jun 1;8(6). doi: 10.1093/gigascience/giz065.
Recent advances in ancient DNA studies, especially in increasing isolated DNA yields and quality, have opened the possibility of analysis of ancient host microbiome. However, such pitfalls as spurious identification of pathogens based on fragmentary data or environmental contamination could lead to incorrect epidaemiological conclusions. Within the Mycobacterium genus, Mycobacterium tuberculosis complex members responsible for tuberculosis share up to ∼99% genomic sequence identity, while other more distantly related Mycobacteria other than M. tuberculosis can be causative agents for pulmonary diseases or soil dwellers. Therefore, reliable determination of species complex is crucial for interpretation of sequencing results.
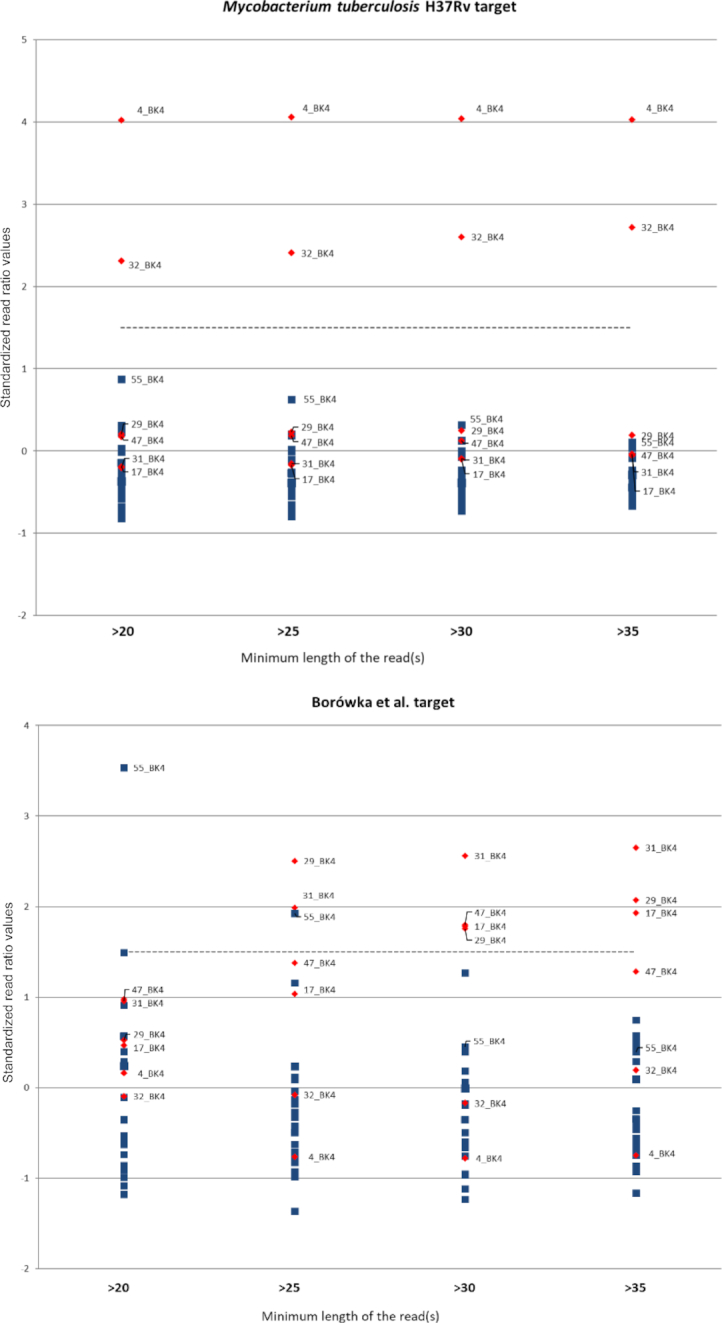
Here we present a novel bioinformatical approach, used for screening of ancient tuberculosis in sequencing data, derived from 28 individuals (dated 4400-4000 and 3100-2900 BC) from central Poland. We demonstrate that cost-effective next-generation screening sequencing data (∼20M reads per sample) could yield enough information to provide statistically supported identification of probable ancient disease cases.
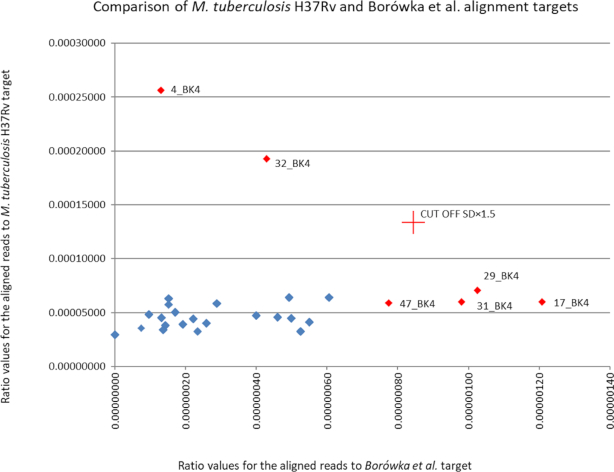
Application of appropriate bioinformatic tools, including an unbiased selection of genomic alignment targets for species specificity, makes it possible to extract valid data from full-sample sequencing results (without subjective targeted enrichment procedures). This approach broadens the potential scope of palaeoepidaemiology both to older, suboptimally preserved samples and to pathogens with difficult intrageneric taxonomy.
最近古 DNA 研究取得了进展,特别是在提高分离 DNA 的产量和质量方面,这使得分析古代宿主微生物组成为可能。然而,基于片段数据或环境污染的假阳性病原体鉴定等陷阱可能导致错误的流行病学结论。在分枝杆菌属中,导致结核病的结核分枝杆菌复合体成员之间的基因组序列同一性高达约 99%,而其他与结核分枝杆菌关系较远的分枝杆菌可能是肺部疾病或土壤栖居者的病原体。因此,可靠地确定种复合体对于解释测序结果至关重要。
我们在此提出了一种新的生物信息学方法,用于筛选来自波兰中部的 28 个人(公元前 4400-4000 年和 3100-2900 年)的测序数据中的古代结核病。我们证明,经济高效的下一代筛选测序数据(每个样本约 2000 万条读长)可以提供足够的信息,以提供支持可能的古代疾病病例的统计鉴定。
应用适当的生物信息学工具,包括对种特异性的基因组比对目标进行无偏见的选择,使得从全样本测序结果中提取有效数据成为可能(无需主观的靶向富集程序)。这种方法拓宽了古流行病学的潜在范围,不仅适用于年代久远、保存不佳的样本,也适用于种内分类学困难的病原体。