Barrell William B, Griffin John N, Harvey Jessica-Lily, Danovi Davide, Beales Philip, Grigoriadis Agamemnon E, Liu Karen J
Centre for Craniofacial and Regenerative Biology, King's College London, London, United Kingdom.
School of Psychology and Neuroscience, University of St Andrews, St Andrews, United Kingdom.
Front Mol Neurosci. 2019 Jun 21;12:139. doi: 10.3389/fnmol.2019.00139. eCollection 2019.
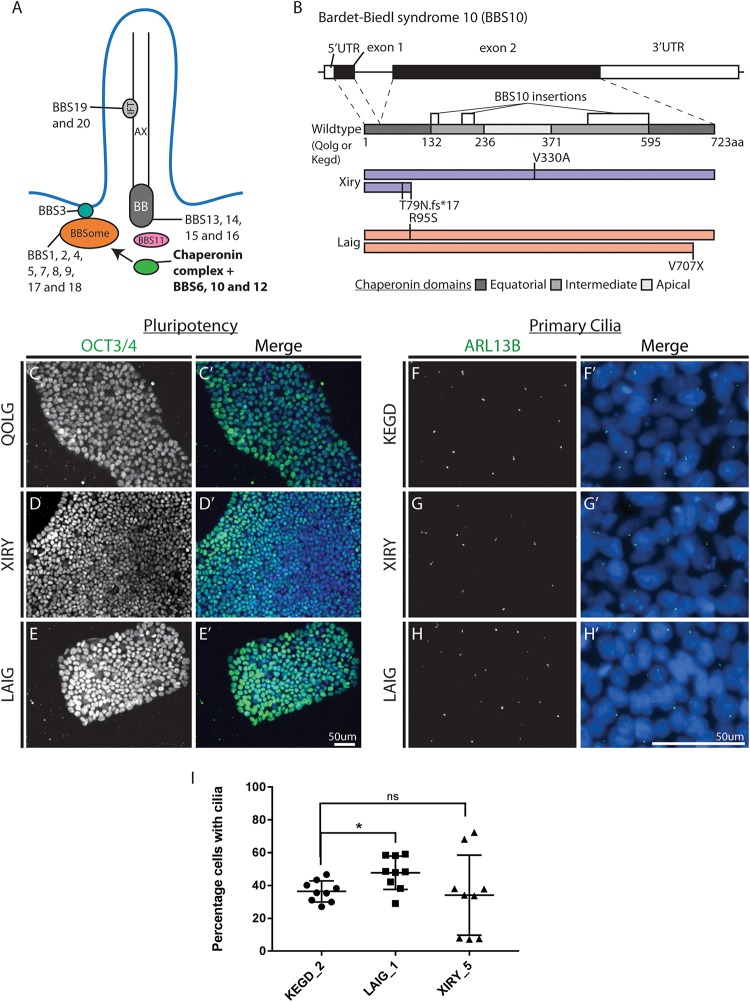
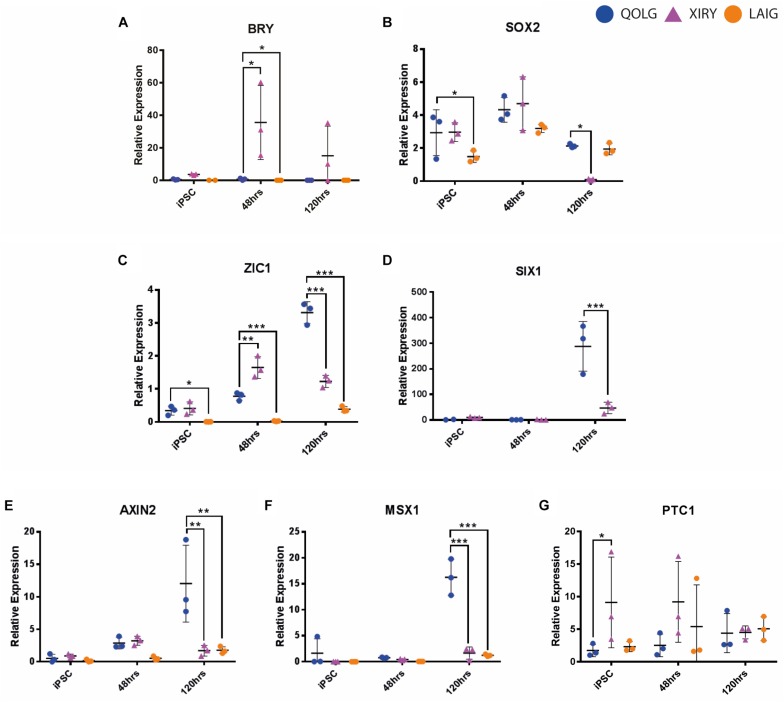
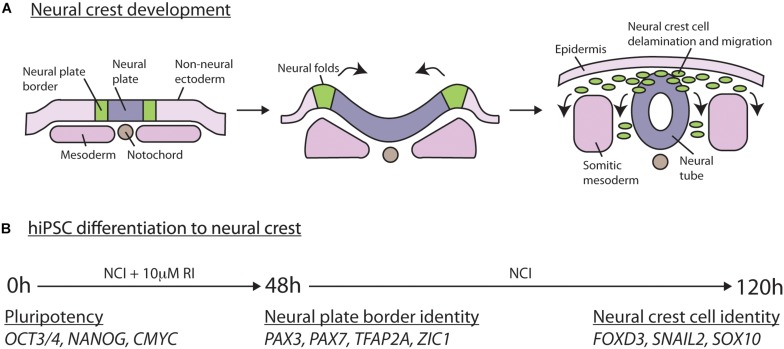
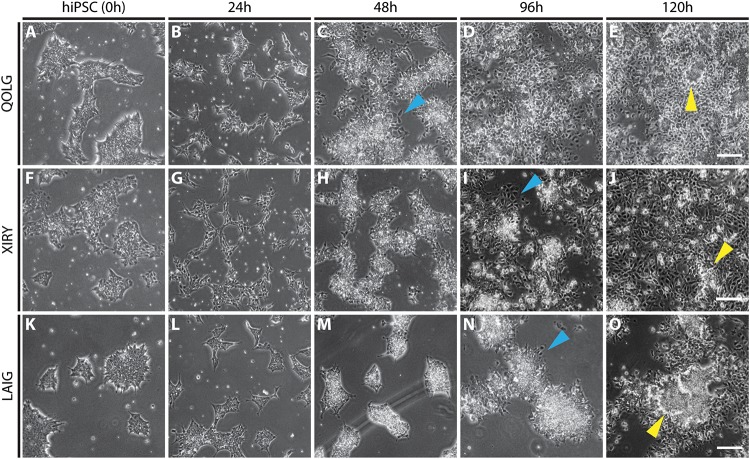
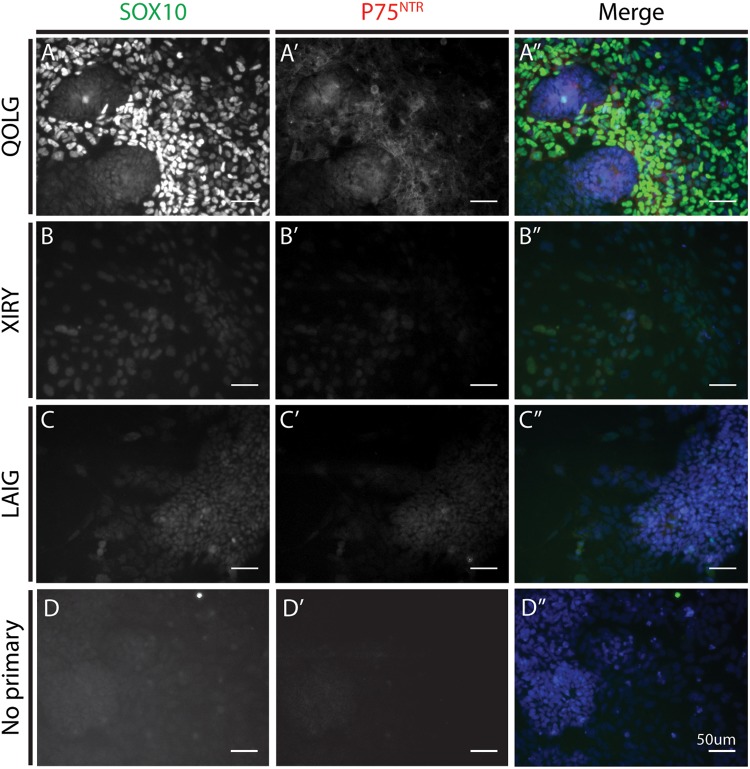
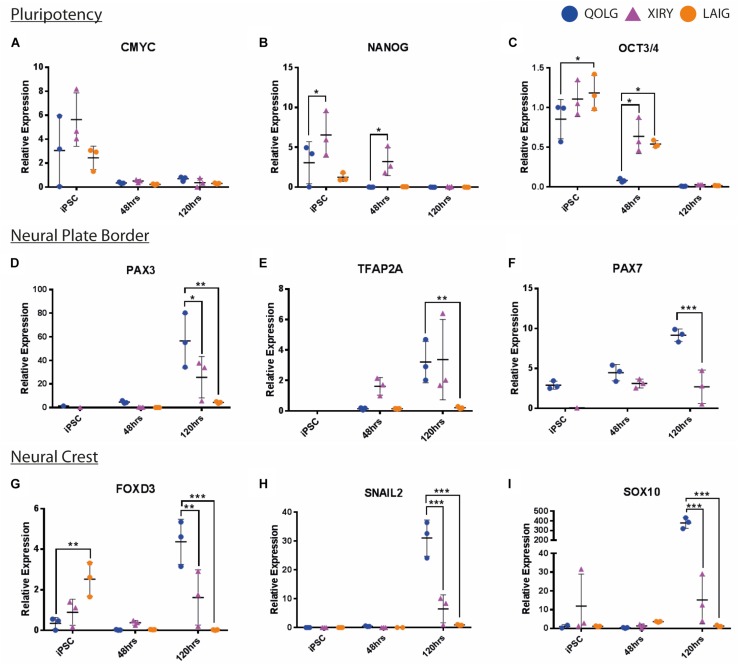
Neural crest cells arise in the embryo from the neural plate border and migrate throughout the body, giving rise to many different tissue types such as bones and cartilage of the face, smooth muscles, neurons, and melanocytes. While studied extensively in animal models, neural crest development and disease have been poorly described in humans due to the challenges in accessing embryonic tissues. In recent years, patient-derived human induced pluripotent stem cells (hiPSCs) have become easier to generate, and several streamlined protocols have enabled robust differentiation of hiPSCs to the neural crest lineage. Thus, a unique opportunity is offered for modeling neurocristopathies using patient specific stem cell lines. In this work, we make use of hiPSCs derived from patients affected by the Bardet-Biedl Syndrome (BBS) ciliopathy. BBS patients often exhibit subclinical craniofacial dysmorphisms that are likely to be associated with the neural crest-derived facial skeleton. We focus on hiPSCs carrying variants in the gene, which encodes a protein forming part of a chaperonin-like complex associated with the cilium. Here, we establish a pipeline for profiling hiPSCs during differentiation toward the neural crest stem cell fate. This can be used to characterize the differentiation properties of the neural crest-like cells. Two different mutant lines showed a reduction in expression of the characteristic neural crest gene expression profile. Further analysis of both mutant lines highlighted the inability of these mutant lines to differentiate toward a neural crest fate, which was also characterized by a decreased WNT and BMP response. Altogether, our study suggests a requirement for wild-type BBS10 in human neural crest development. In the long term, approaches such as the one we describe will allow direct comparison of disease-specific cell lines. This will provide valuable insights into the relationships between genetic background and heterogeneity in cellular models. The possibility of integrating laboratory data with clinical phenotypes will move us toward precision medicine approaches.
神经嵴细胞在胚胎中起源于神经板边界,并迁移至全身,产生许多不同的组织类型,如面部的骨骼和软骨、平滑肌、神经元和黑素细胞。虽然在动物模型中对神经嵴进行了广泛研究,但由于获取胚胎组织存在挑战,神经嵴发育和疾病在人类中的描述却很少。近年来,患者来源的人诱导多能干细胞(hiPSC)变得更容易生成,并且一些简化的方案能够使hiPSC强有力地分化为神经嵴谱系。因此,利用患者特异性干细胞系对神经嵴病进行建模提供了一个独特的机会。在这项工作中,我们利用了源自患有巴德-比埃尔综合征(BBS)纤毛病患者的hiPSC。BBS患者通常表现出亚临床颅面畸形,这可能与神经嵴衍生的面部骨骼有关。我们关注携带该基因变体的hiPSC,该基因编码一种构成与纤毛相关的伴侣蛋白样复合物一部分的蛋白质。在这里,我们建立了一个在向神经嵴干细胞命运分化过程中对hiPSC进行分析的流程。这可用于表征神经嵴样细胞的分化特性。两个不同的突变系显示特征性神经嵴基因表达谱的表达降低。对这两个突变系的进一步分析突出了这些突变系无法向神经嵴命运分化,这也表现为WNT和BMP反应降低。总之,我们的研究表明野生型BBS10在人类神经嵴发育中是必需的。从长远来看,我们所描述的方法将允许对疾病特异性细胞系进行直接比较。这将为细胞模型中遗传背景与异质性之间的关系提供有价值的见解。将实验室数据与临床表型整合的可能性将推动我们走向精准医学方法。