State Key Laboratory of Tea Plant Biology and Utilization, Anhui Agricultural University, Hefei, 230036, China.
BGI-Shenzhen, Shenzhen, 518083, China.
Sci Data. 2019 Jul 15;6(1):122. doi: 10.1038/s41597-019-0127-1.
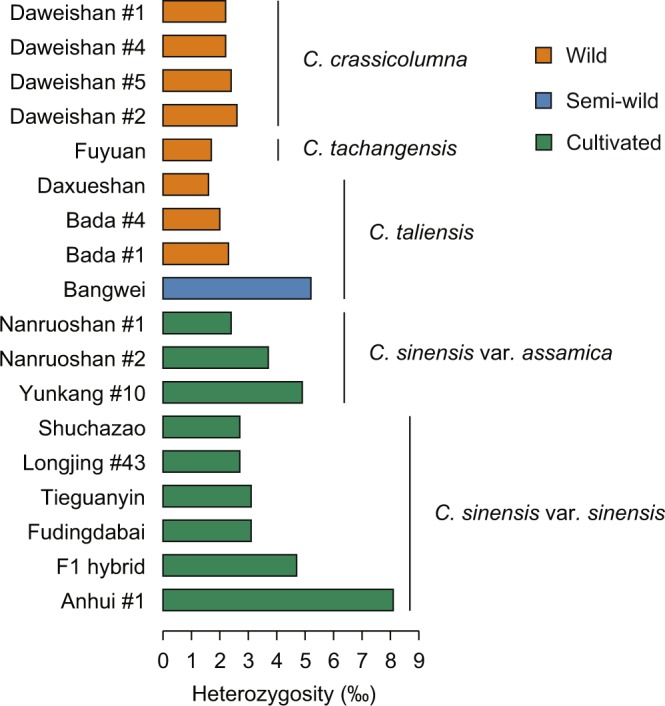
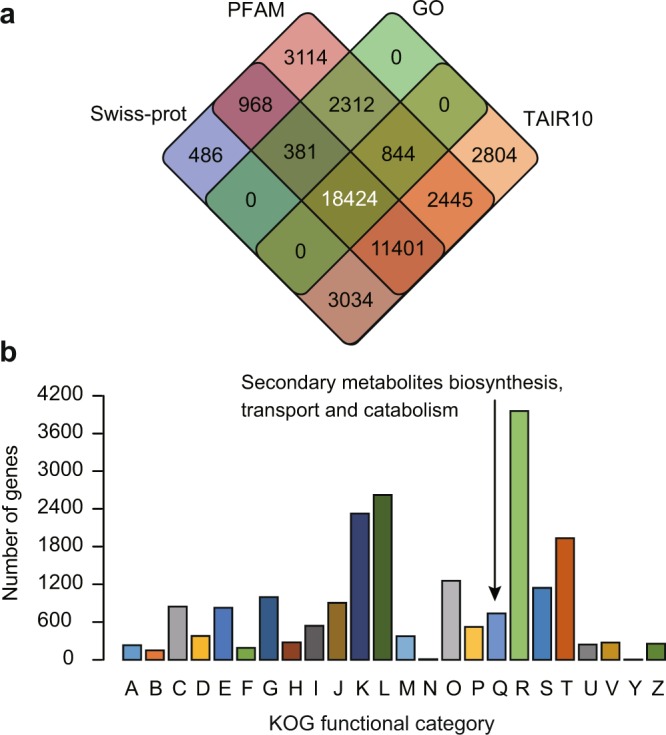
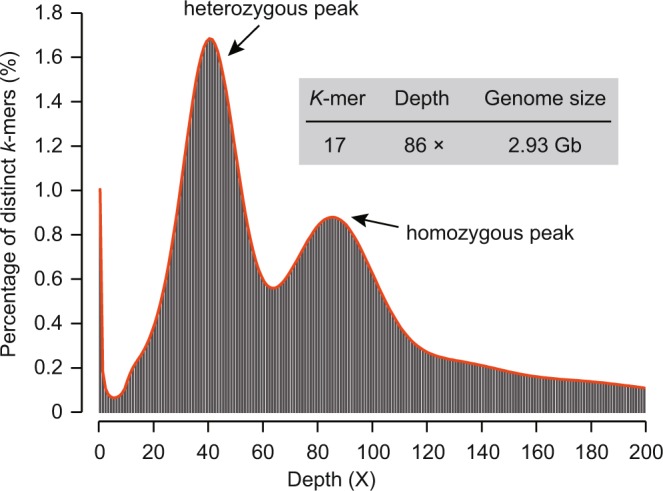
Tea is a globally consumed non-alcohol beverage with great economic importance. However, lack of the reference genome has largely hampered the utilization of precious tea plant genetic resources towards breeding. To address this issue, we previously generated a high-quality reference genome of tea plant using Illumina and PacBio sequencing technology, which produced a total of 2,124 Gb short and 125 Gb long read data, respectively. A hybrid strategy was employed to assemble the tea genome that has been publicly released. We here described the data framework used to generate, annotate and validate the genome assembly. Besides, we re-predicted the protein-coding genes and annotated their putative functions using more comprehensive omics datasets with improved training models. We reassessed the assembly and annotation quality using the latest version of BUSCO. These data can be utilized to develop new methodologies/tools for better assembly of complex genomes, aid in finding of novel genes, variations and evolutionary clues associated with tea quality, thus help to breed new varieties with high yield and better quality in the future.
茶是一种全球消费的非酒精饮料,具有重要的经济意义。然而,由于缺乏参考基因组,极大地阻碍了宝贵的茶树遗传资源在培育方面的利用。为了解决这个问题,我们之前使用 Illumina 和 PacBio 测序技术生成了一个高质量的茶树参考基因组,分别产生了总计 2124GB 的短读和 125GB 的长读数据。我们采用了一种混合策略来组装已经公开的茶树基因组。在这里,我们描述了用于生成、注释和验证基因组组装的数据集框架。此外,我们使用更全面的组学数据集和改进的训练模型重新预测了蛋白质编码基因,并注释了它们的可能功能。我们使用最新版本的 BUSCO 重新评估了组装和注释的质量。这些数据可用于开发新的方法/工具,以更好地组装复杂基因组,帮助发现与茶叶质量相关的新基因、变异和进化线索,从而有助于未来培育出高产、优质的新品种。