Christopoulos Stavros-Richard G, Kuganathan Navaratnarajah, Chroneos Alexander
Faculty of Engineering, Environment and Computing, Coventry University, Priory Street, Coventry, CV1 5FB, United Kingdom.
Department of Materials, Imperial College London, London, SW7 2AZ, United Kingdom.
Sci Rep. 2019 Jul 26;9(1):10849. doi: 10.1038/s41598-019-47385-0.
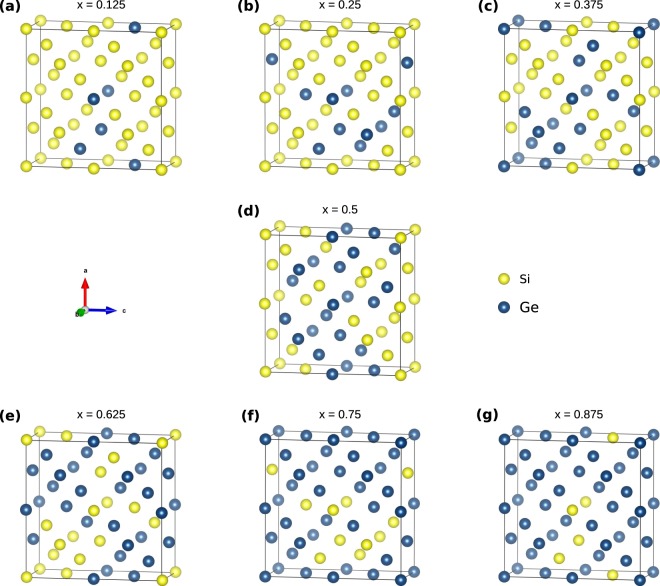
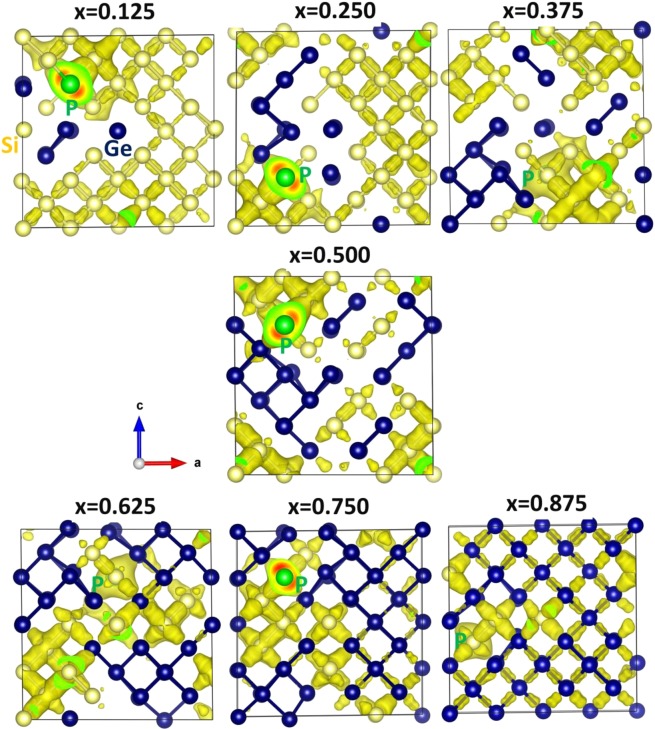
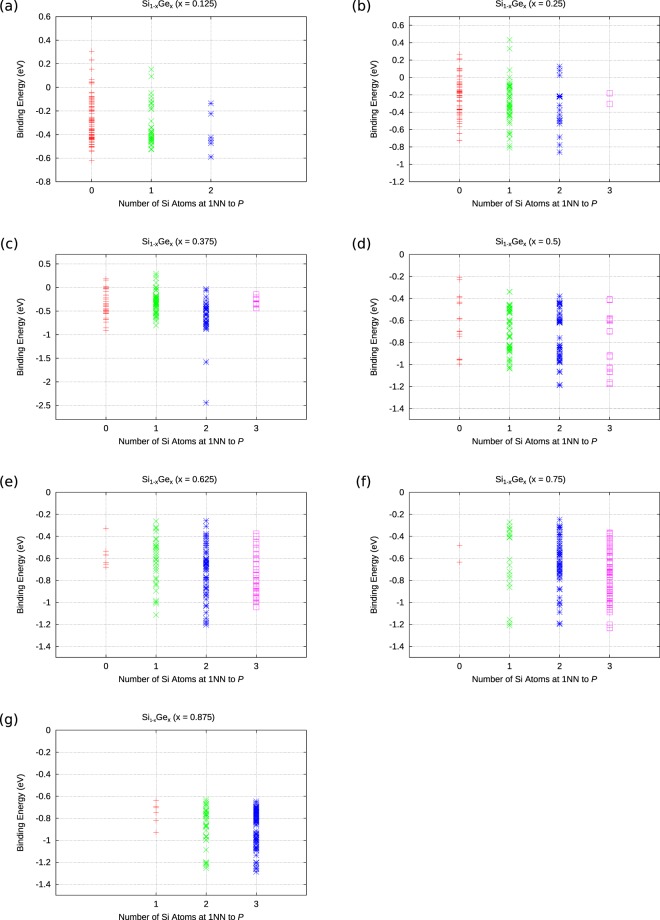
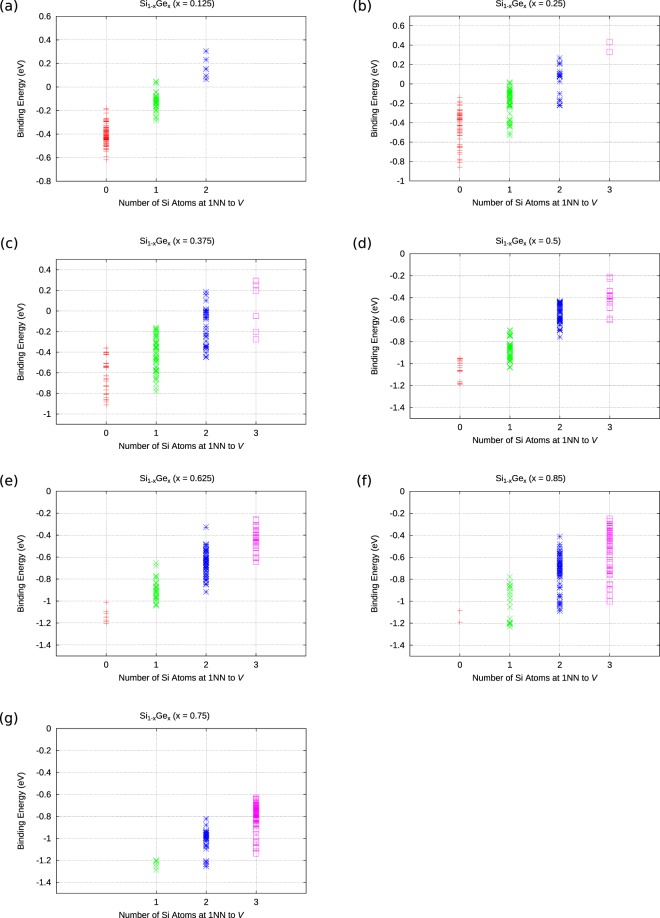
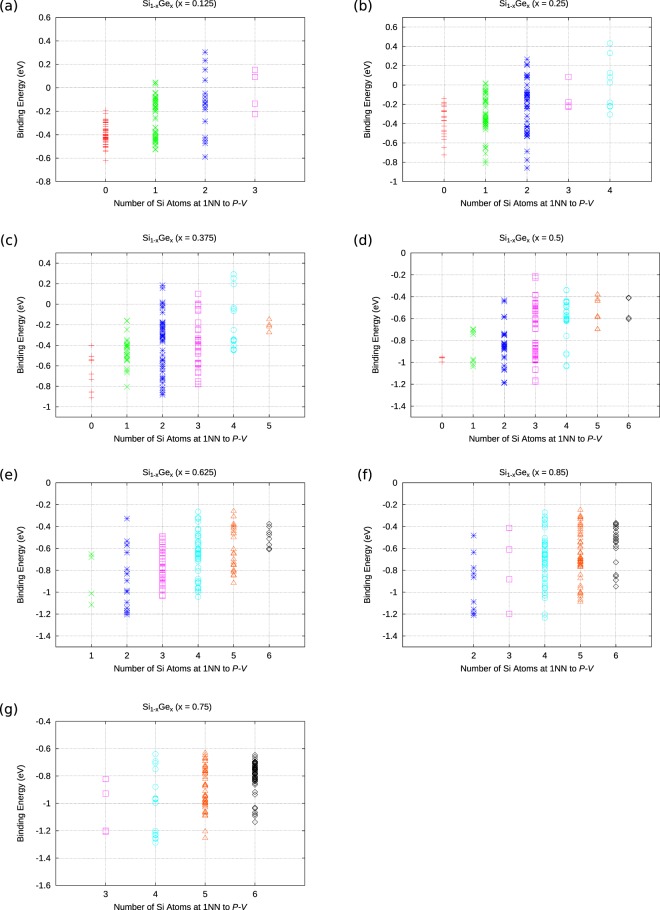
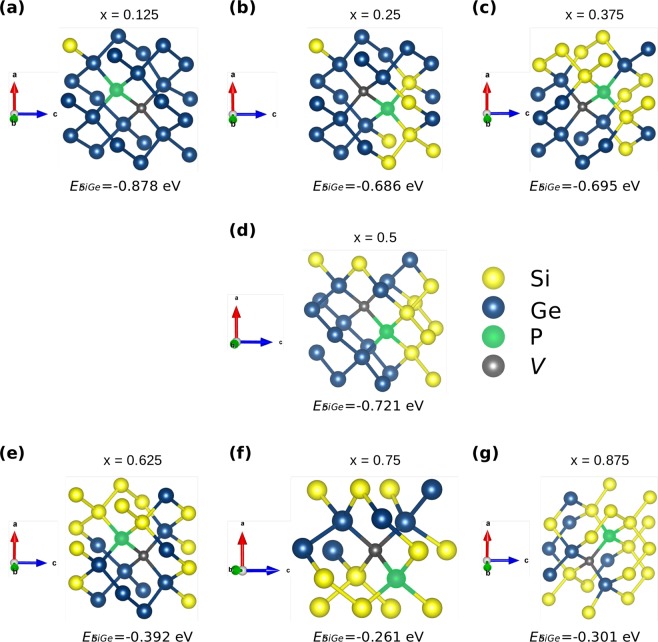
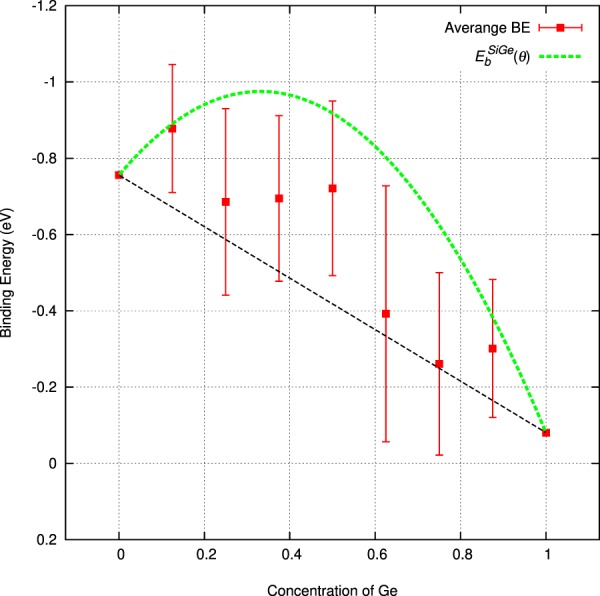
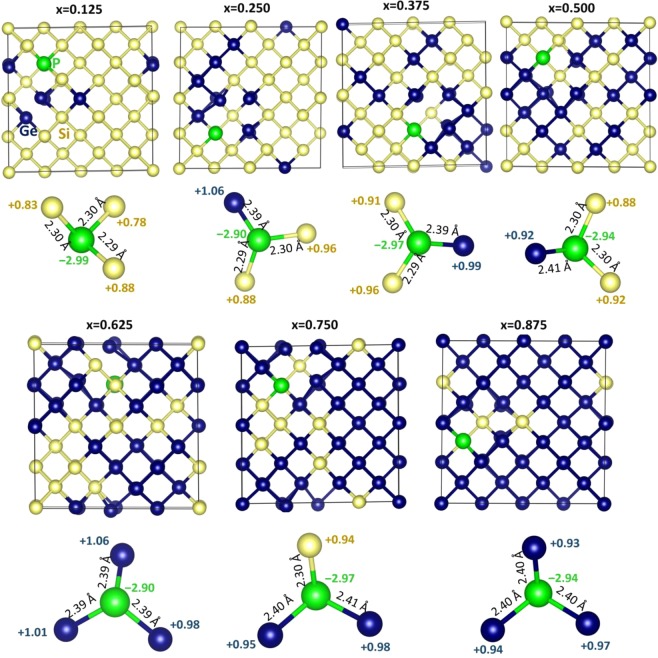
The energetics of the defect chemistry and processes in semiconducting alloys is both technologically and theoretically significant. This is because defects in semiconductors are critical to tune their electronic properties. These processes are less well understood in random semiconductor alloys such as silicon germanium as compared to elementary semiconductors (for example silicon). To model the random silicon germanium alloy we have employed density functional theory calculations in conjunction with the special quasirandom structures model for different compositions. Here we show that, the energetics of substitutional phosphorous-vacancy pairs (E-centres) in SiGe alloys vary greatly with respect to the local Ge concentration and the composition of the alloy. The most energetically favourable E-centres have a Ge atom as a nearest neighbour, whereas the dependence of the binding energy of the E-centres with respect to alloy composition is non-linear.
半导体合金中缺陷化学和过程的能量学在技术和理论上都具有重要意义。这是因为半导体中的缺陷对于调节其电子特性至关重要。与单质半导体(例如硅)相比,在诸如硅锗之类的随机半导体合金中,这些过程的理解尚不充分。为了对随机硅锗合金进行建模,我们结合不同成分的特殊准随机结构模型,采用了密度泛函理论计算。在此我们表明,硅锗合金中替代磷空位对(E中心)的能量学随局部锗浓度和合金成分的变化很大。能量上最有利的E中心以一个锗原子作为最近邻,而E中心的结合能相对于合金成分的依赖性是非线性的。