Department of Chemistry , Massachusetts Institute of Technology , Cambridge , Massachusetts 02139 , United States.
J Am Chem Soc. 2019 Sep 11;141(36):14160-14167. doi: 10.1021/jacs.9b04981. Epub 2019 Aug 29.
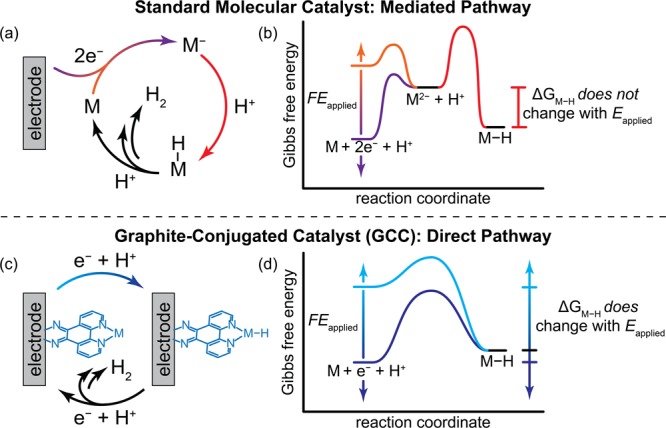
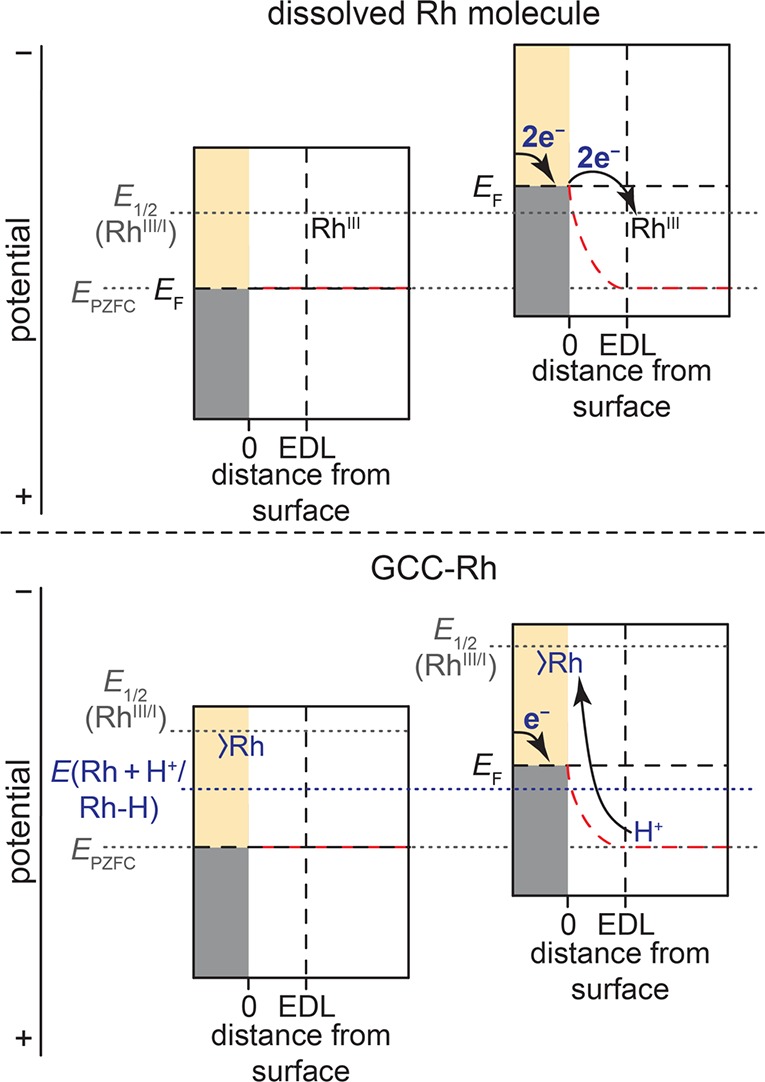
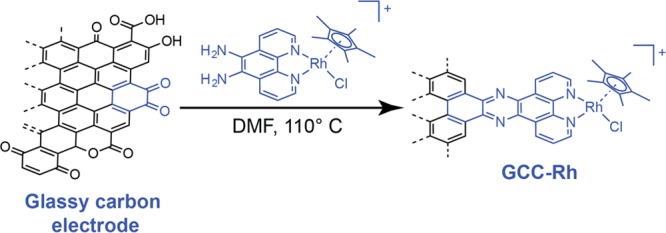
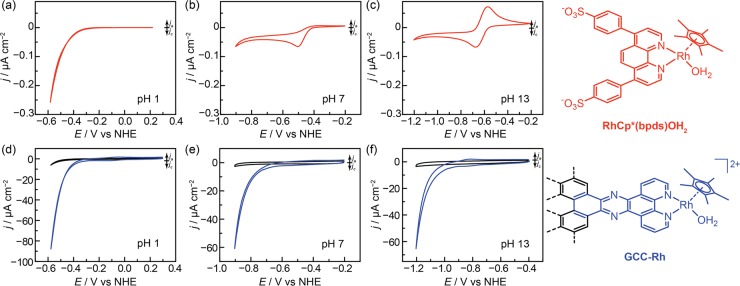
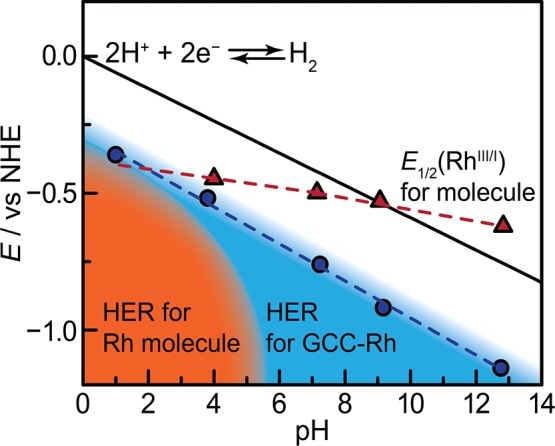
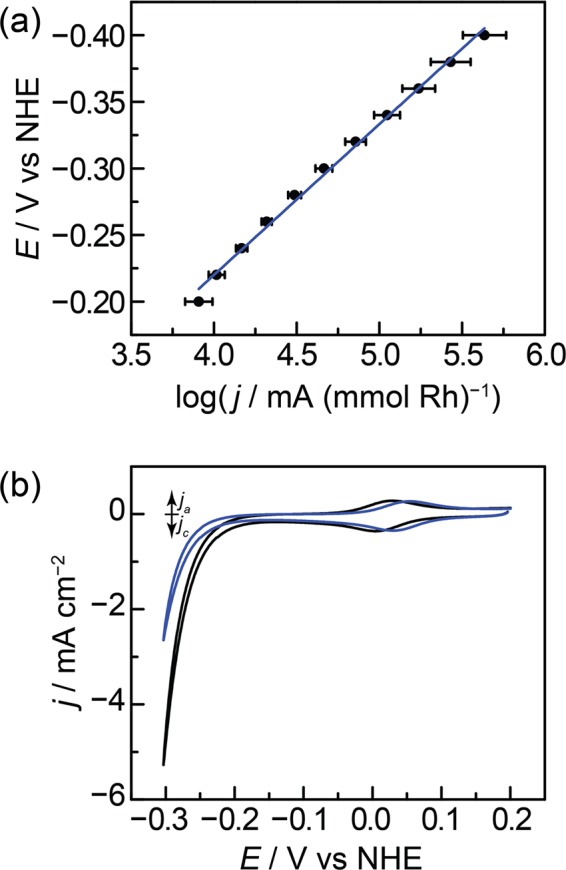
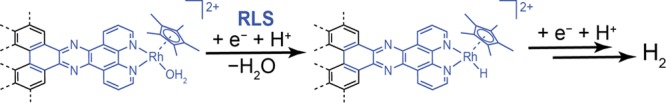
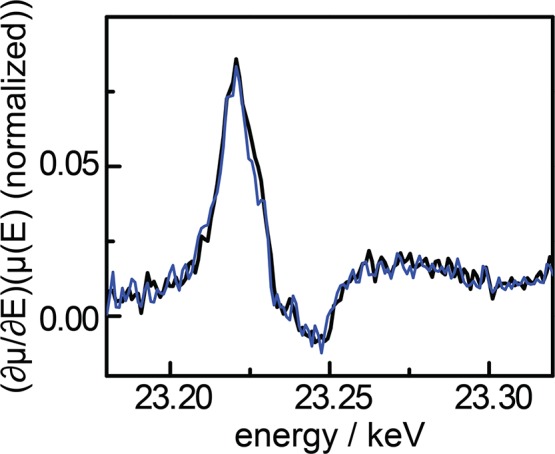
The efficient interconversion of electrical and chemical energy requires the intimate coupling of electrons and small-molecule substrates at catalyst active sites. In molecular electrocatalysis, the molecule acts as a redox mediator which typically undergoes oxidation or reduction in a separate step from substrate activation. These mediated pathways introduce a high-energy intermediate, cap the driving force for substrate activation at the reduction potential of the molecule, and impede access to high rates at low overpotentials. Here we show that electronically coupling a molecular hydrogen evolution catalyst to a graphitic electrode eliminates stepwise pathways and forces concerted electron transfer and proton binding. Electrochemical and X-ray absorption spectroscopy data establish that hydrogen evolution catalysis at the graphite-conjugated Rh molecule proceeds without first reducing the metal center. These results have broad implications for the molecular-level design of energy conversion catalysts.
电和化学能的高效转换需要电子和小分子底物在催化剂活性位点上的紧密偶联。在分子电催化中,分子作为氧化还原介体,通常在与底物活化分开的步骤中发生氧化或还原。这些介导途径引入了一个高能中间体,将底物活化的驱动力限制在分子的还原电位,并阻碍了在低过电势下达到高速率的能力。在这里,我们表明,将分子氢析出催化剂与石墨电极电子耦合消除了分步途径,并强制协同电子转移和质子结合。电化学和 X 射线吸收光谱数据表明,石墨共轭 Rh 分子的析氢催化作用并不首先还原金属中心。这些结果对能量转换催化剂的分子水平设计具有广泛的意义。