Department of Environmental Science, Aarhus University RISØ Campus, Frederiksborgvej 399, 4000 Roskilde, Denmark.
AZTI, Herrera Kaia, Portualdea z/g, 20110 Pasaia, Basque Country, Spain.
Gigascience. 2019 Aug 1;8(8). doi: 10.1093/gigascience/giz096.
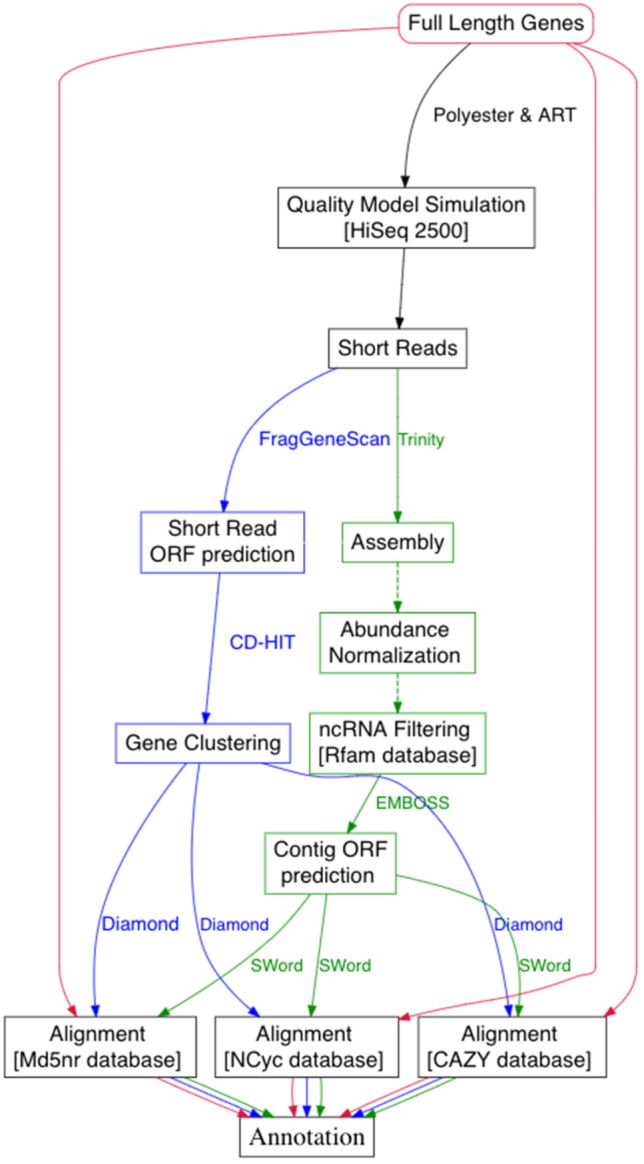
Metatranscriptomics has been used widely for investigation and quantification of microbial communities' activity in response to external stimuli. By assessing the genes expressed, metatranscriptomics provides an understanding of the interactions between different major functional guilds and the environment. Here, we present a de novo assembly-based Comparative Metatranscriptomics Workflow (CoMW) implemented in a modular, reproducible structure. Metatranscriptomics typically uses short sequence reads, which can either be directly aligned to external reference databases ("assembly-free approach") or first assembled into contigs before alignment ("assembly-based approach"). We also compare CoMW (assembly-based implementation) with an assembly-free alternative workflow, using simulated and real-world metatranscriptomes from Arctic and temperate terrestrial environments. We evaluate their accuracy in precision and recall using generic and specialized hierarchical protein databases.
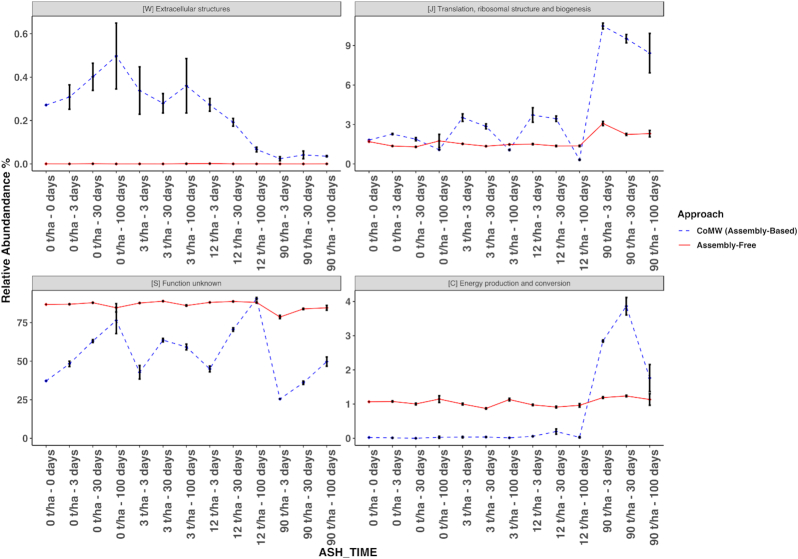
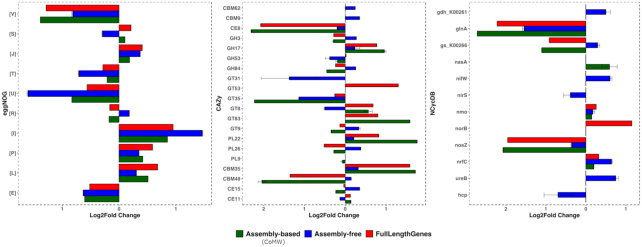
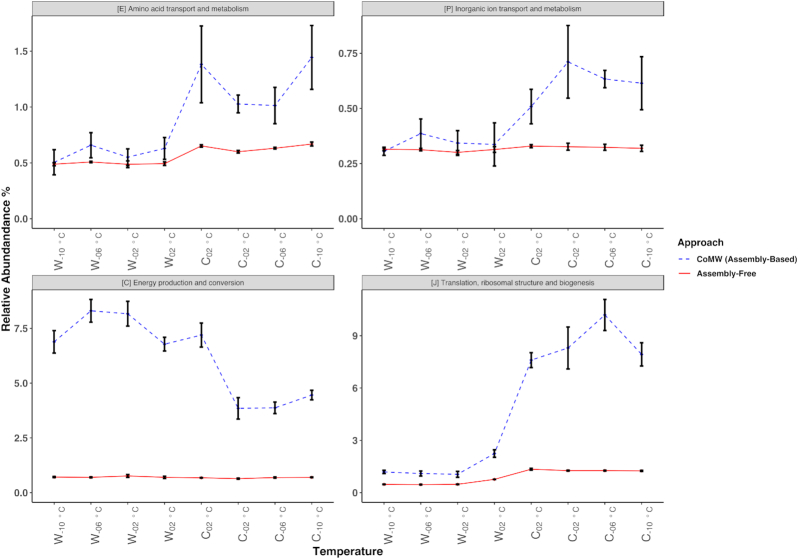
CoMW provided significantly fewer false-positive results, resulting in more precise identification and quantification of functional genes in metatranscriptomes. Using the comprehensive database M5nr, the assembly-based approach identified genes with only 0.6% false-positive results at thresholds ranging from inclusive to stringent compared with the assembly-free approach, which yielded up to 15% false-positive results. Using specialized databases (carbohydrate-active enzyme and nitrogen cycle), the assembly-based approach identified and quantified genes with 3-5 times fewer false-positive results. We also evaluated the impact of both approaches on real-world datasets.
We present an open source de novo assembly-based CoMW. Our benchmarking findings support assembling short reads into contigs before alignment to a reference database because this provides higher precision and minimizes false-positive results.
宏转录组学已广泛应用于研究和量化微生物群落对外界刺激的活性。通过评估表达的基因,宏转录组学可以了解不同主要功能类群与环境之间的相互作用。在这里,我们提出了一种基于从头组装的比较宏转录组学工作流程(CoMW),该工作流程采用模块化、可重复的结构实现。宏转录组学通常使用短序列读段,可以直接与外部参考数据库对齐(“无组装方法”),也可以在对齐之前组装成 contigs(“基于组装的方法”)。我们还使用来自北极和温带陆地环境的模拟和真实宏转录组数据,比较了 CoMW(基于组装的实现)与无组装的替代工作流程。我们使用通用和专门的层次蛋白质数据库来评估它们在精度和召回率方面的准确性。
CoMW 提供的假阳性结果明显较少,从而更精确地识别和定量宏转录组中的功能基因。使用综合数据库 M5nr,与无组装方法相比,基于组装的方法在从包容到严格的阈值范围内识别基因的假阳性结果仅为 0.6%,而无组装方法的假阳性结果高达 15%。使用专门的数据库(碳水化合物活性酶和氮循环),基于组装的方法识别和定量基因的假阳性结果减少了 3-5 倍。我们还评估了这两种方法对真实数据集的影响。
我们提出了一种开源的基于从头组装的 CoMW。我们的基准测试结果支持在将短读段与参考数据库对齐之前将其组装成 contigs,因为这可以提供更高的精度并最大限度地减少假阳性结果。