Barbosa-Gouveia Sofia, González-Vioque Emiliano, Borges Filipa, Gutiérrez-Solana Luis, Wintjes Liesbeth, Kappen Antonia, van den Heuvel Lambert, Leis Rosaura, Rodenburg Richard, Couce María Luz
Diagnosis and Treatment of Congenital Metabolic Diseases Unit (UDyTEMC), Department of Pediatrics, Clinical University Hospital of Santiago de Compostela, 15706 Santiago de Compostela, Spain.
Faculty of Medicine, University of Santiago de Compostela, 15706 Santiago de Compostela, Spain.
J Clin Med. 2019 Aug 20;8(8):1262. doi: 10.3390/jcm8081262.
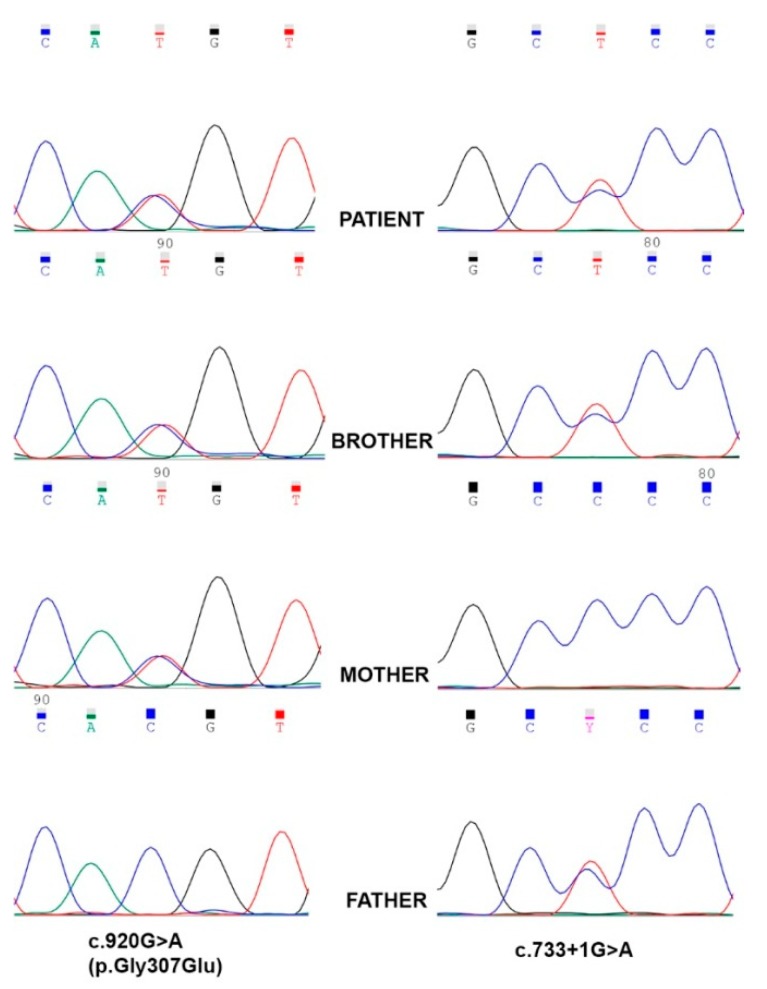
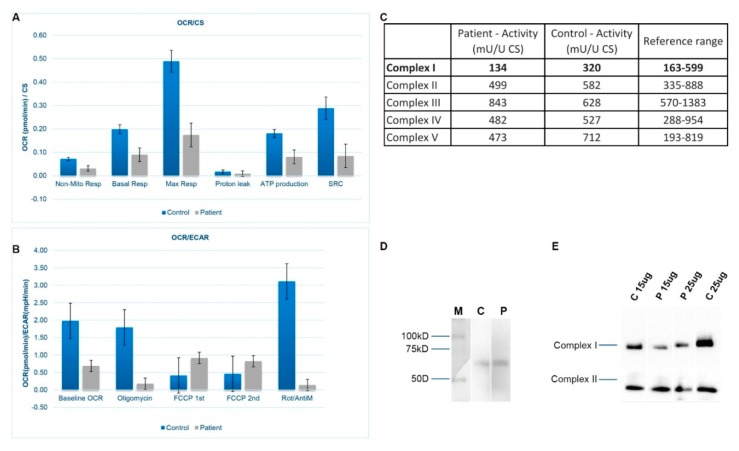
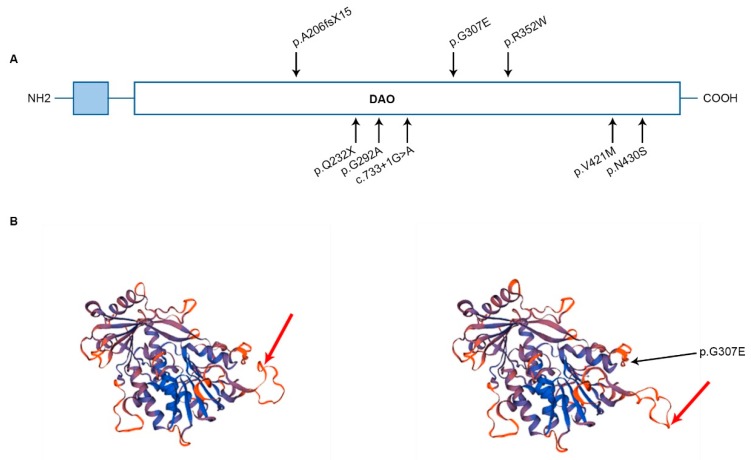
Complex I (nicotinamide adenine dinucleotide (NADH): ubiquinone oxidoreductase) is the largest complex of the mitochondrial oxidative phosphorylation system (OXPHOS) system. Forty-four subunits encoded in nuclear and mitochondrial genomes compose this multiprotein complex, its assembly being a highly complex process involving at least 15 additional nuclear encoded assembly factors. Complex I deficiency is a mitochondrial disorder usually associated with early-onset severe multisystem disorders characterized by highly variable clinical manifestations. Flavin adenine dinucleotide (FAD)-dependent oxidoreductase domain-containing protein 1 (FOXRED1) is a complex I assembly factor. To date, only five patients with mitochondrial complex I deficiency due to mutations in have been characterized. Here, we describe a child with ataxia, epilepsy and psychomotor developmental delay carrying two heterozygous variants, c.920G>A (p.Gly307Glu) and c.733+1G>A. We demonstrate the molecular mechanism supporting the pathogenicity of the FOXRED1 variants, showing a clear deficiency of complex I activity. The reduction in the steady-state level of complex I holoenzyme in patient fibroblasts, confirmed the pathogenicity of the variants and showed the molecular mechanism behind their pathogenicity. A comparison of the clinical presentation of the index case with the previously described cases allowed deepening our knowledge about the clinical variability associated with defects.
复合体I(烟酰胺腺嘌呤二核苷酸(NADH):泛醌氧化还原酶)是线粒体氧化磷酸化系统(OXPHOS)中最大的复合体。由核基因组和线粒体基因组编码的44个亚基组成了这个多蛋白复合体,其组装是一个高度复杂的过程,至少涉及15个额外的核编码组装因子。复合体I缺乏是一种线粒体疾病,通常与早发性严重多系统疾病相关,其临床表现高度可变。含黄素腺嘌呤二核苷酸(FAD)的依赖氧化还原酶结构域蛋白1(FOXRED1)是复合体I的一个组装因子。迄今为止,仅对5例因 突变导致线粒体复合体I缺乏的患者进行了特征描述。在此,我们描述了一名患有共济失调、癫痫和精神运动发育迟缓的儿童携带两个杂合 变体,即c.920G>A(p.Gly307Glu)和c.733+1G>A。我们证明了支持FOXRED1变体致病性的分子机制,显示出复合体I活性明显缺乏。患者成纤维细胞中复合体I全酶稳态水平的降低,证实了这些变体的致病性,并揭示了其致病性背后的分子机制。将该索引病例的临床表现与先前描述的病例进行比较,有助于加深我们对与 缺陷相关的临床变异性的认识。