Hu Chaoping, Xu Qiong, Shen Jin, Wang Yi
Department of Neurology, Children's Hospital of Fudan University, Shanghai, China.
Department of Child Health Care, Children's Hospital of Fudan University, Shanghai, China.
Front Neurol. 2021 Feb 3;12:633397. doi: 10.3389/fneur.2021.633397. eCollection 2021.
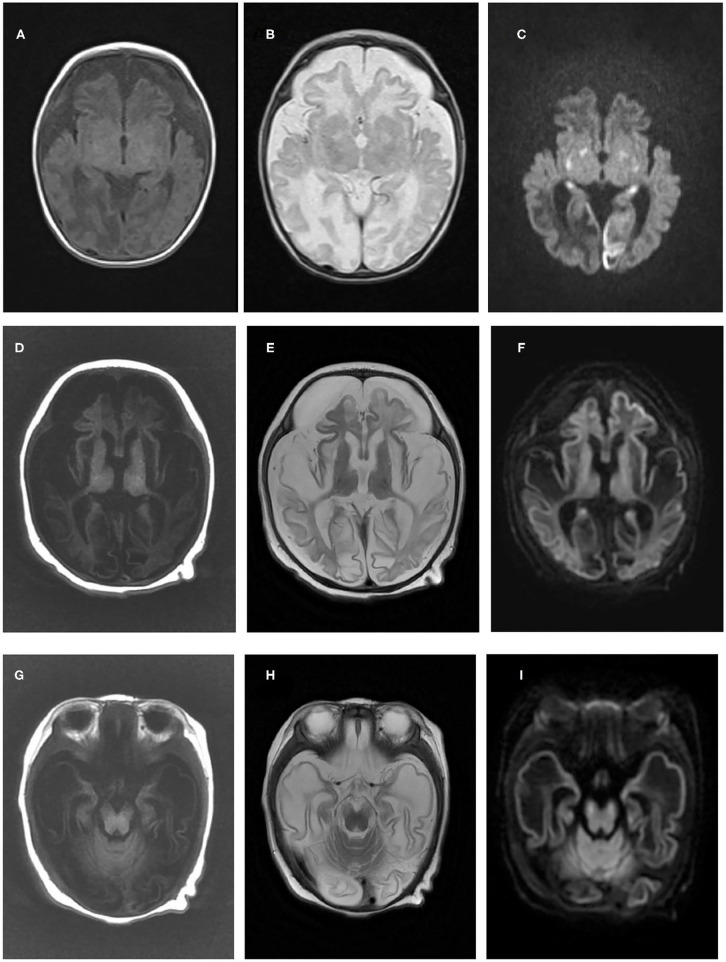
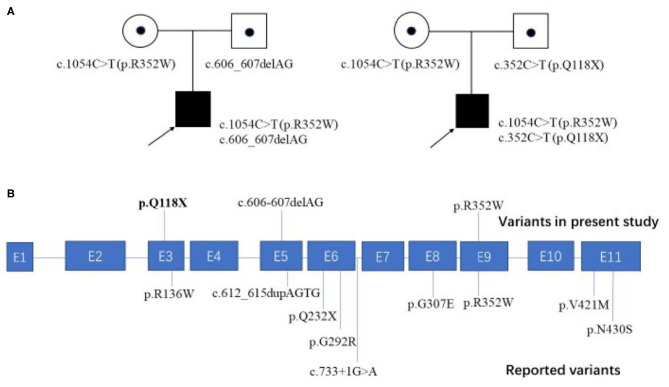
As one of the assembly factors of complex I in the mitochondrial respiratory chain, FOXRED1 plays an important role in mitochondrial function. However, only a few patients with mitochondrial encephalopathy due to FOXRED1 defects have been reported. Two Chinese patients with mitochondrial encephalopathy due to mutations in were identified through trio whole-exome sequencing. The clinical presentation, laboratory data, brain imaging findings, and genetic results were collected and reviewed. All previously reported cases with -related mitochondrial encephalopathy were collected using a PubMed search, and their data were reviewed. Two patients presented with severe neurodevelopmental delay, epilepsy, high lactic acid levels, and remarkable diffuse brain atrophy and polycystic encephalomalacia during early infancy. Trio whole-exome sequencing revealed compound heterozygous variants in both patients: one case harbored a c.606_607delAG frameshift variant and a c.1054C>T (p.R352W) variant. At the same time, the other carried a novel c.352C>T (p.Q118X) variant and a reported c.1054C>T (p.R352W) variant. To date, nine patients have been reported with defects, including our two cases. The most common presentations were neurodevelopment delay (100%), epilepsy (80%), poor feeding (30%), and vision loss (20%). Multisystem involvement comprised cardiovascular dysfunction (30%), abnormal liver function (20%), and hypoglycemia (10%). The neuroimaging results ranged from normal to severe cerebral atrophy and polycystic encephalomalacia in early infancy. Eleven pathogenic variants in FOXRED1 have been reported, comprising six missense variants, two non-sense variants, two frameshift variants, and one splice variant; among these the c.1054C>T (p.R352W) and c.612_615dupAGTG (p.A206SfsX15) variants are more common. -related mitochondrial disorders have high clinical and genetic heterogeneity. Our study expanded the clinical and genetic spectrum of defects. Early infantile onset and progressive encephalopathy are the most common clinical presentations, while the variants c.1054C>T (p.R352W) and c.612_615dupAGTG (p.A206SfsX15) may be critical founder mutations.
作为线粒体呼吸链中复合物I的组装因子之一,FOXRED1在线粒体功能中发挥着重要作用。然而,仅有少数因FOXRED1缺陷导致线粒体脑病的患者被报道。通过三联体全外显子测序鉴定出两名因 突变导致线粒体脑病的中国患者。收集并回顾了他们的临床表现、实验室数据、脑成像结果及基因检测结果。通过PubMed检索收集了所有先前报道的与 相关的线粒体脑病病例,并对其数据进行了回顾。两名患者在婴儿早期均出现严重的神经发育迟缓、癫痫、高乳酸血症,以及明显的弥漫性脑萎缩和多囊性脑软化。三联体全外显子测序显示两名患者均为复合杂合变异:一例携带c.606_607delAG移码变异和c.1054C>T(p.R352W)变异;另一例携带一个新的c.352C>T(p.Q118X)变异和一个已报道的c.1054C>T(p.R352W)变异。迄今为止,包括我们的两例病例在内,已有9例患者被报道存在 缺陷。最常见的表现为神经发育迟缓(100%)、癫痫(80%)、喂养困难(30%)和视力丧失(20%)。多系统受累包括心血管功能障碍(30%)、肝功能异常(20%)和低血糖(10%)。神经影像学结果从正常到婴儿早期严重的脑萎缩和多囊性脑软化不等。已报道了11种FOXRED1的致病变异,包括6种错义变异、2种无义变异、2种移码变异和1种剪接变异;其中c.1054C>T(p.R352W)和c.612_615dupAGTG(p.A206SfsX15)变异较为常见。 相关的线粒体疾病具有高度的临床和遗传异质性。我们的研究扩展了 缺陷的临床和遗传谱。婴儿早期起病和进行性脑病是最常见的临床表现,而c.1054C>T(p.R352W)和c.612_615dupAGTG(p.A206SfsX15)变异可能是关键的奠基者突变。 (注:原文中部分内容缺失具体基因名称,用 表示)