Dong Xuesi, Zhang Ruyang, He Jieyu, Lai Linjing, Alolga Raphael N, Shen Sipeng, Zhu Ying, You Dongfang, Lin Lijuan, Chen Chao, Zhao Yang, Duan Weiwei, Su Li, Shafer Andrea, Salama Moran, Fleischer Thomas, Bjaanæs Maria Moksnes, Karlsson Anna, Planck Maria, Wang Rui, Staaf Johan, Helland Åslaug, Esteller Manel, Wei Yongyue, Chen Feng, Christiani David C
Department of Epidemiology and Biostatistics, School of Public Health, Southeast University, Nanjing 210009, China.
Department of Biostatistics, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing 211166, China.
Aging (Albany NY). 2019 Aug 21;11(16):6312-6335. doi: 10.18632/aging.102189.
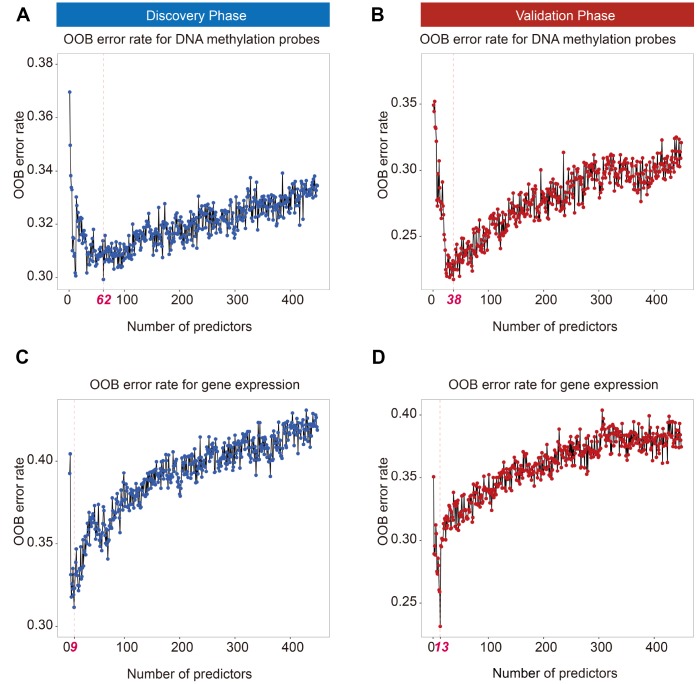
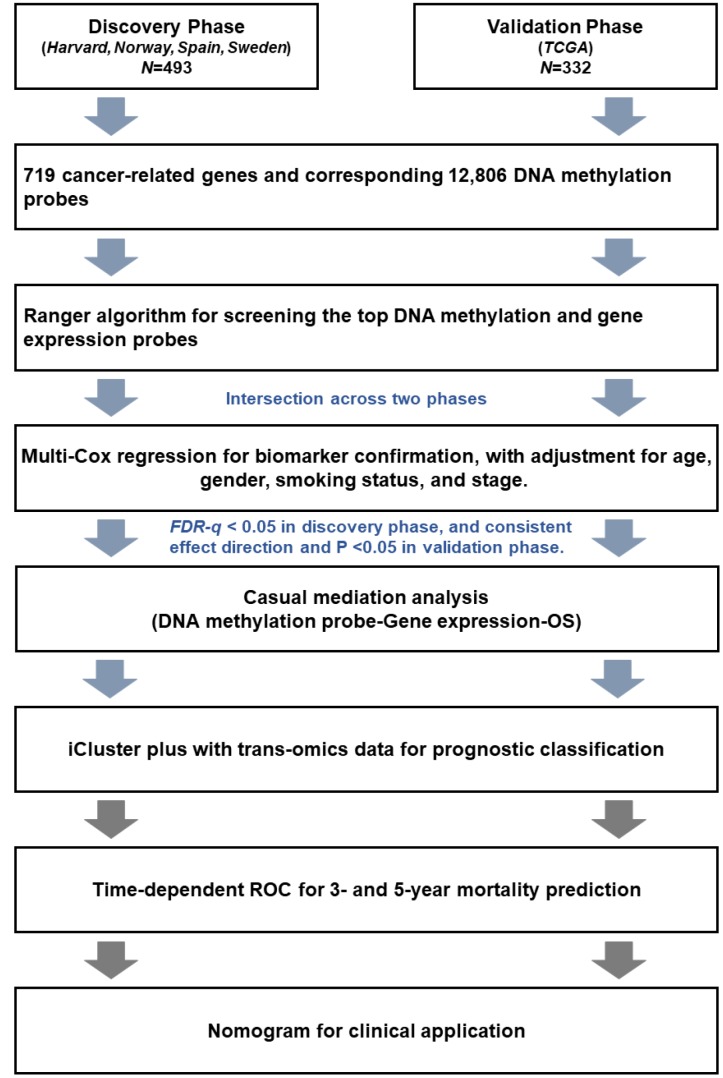
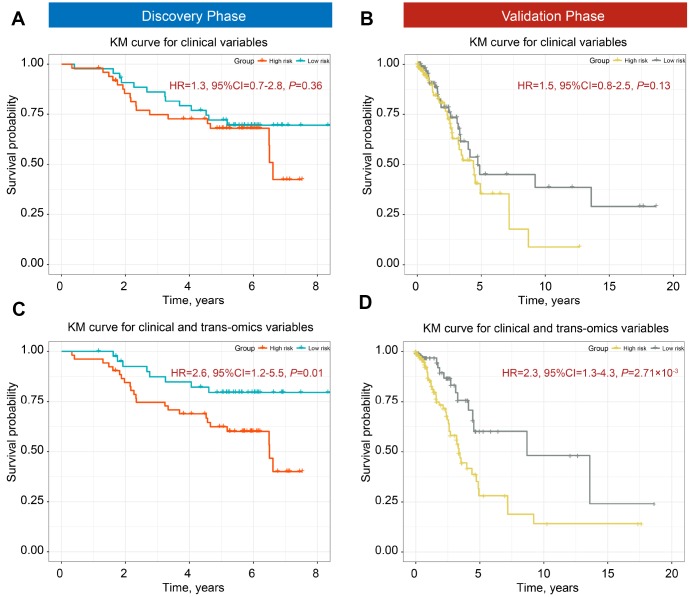
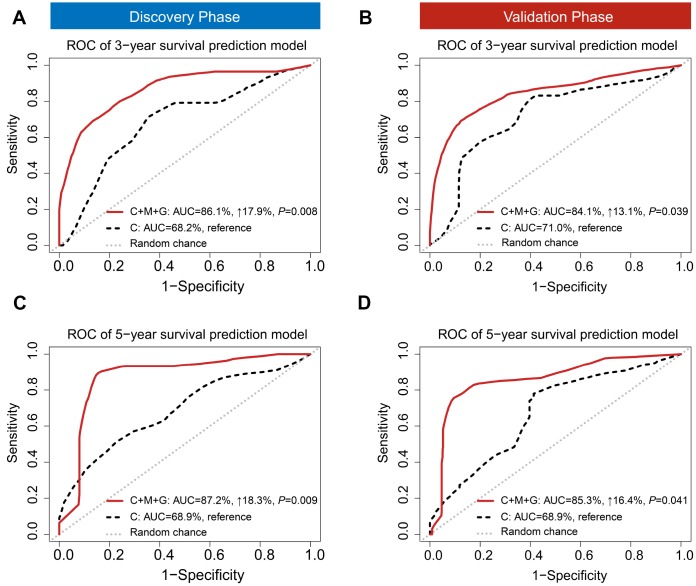
Limited studies have focused on developing prognostic models with trans-omics biomarkers for early-stage lung adenocarcinoma (LUAD). We performed integrative analysis of clinical information, DNA methylation, and gene expression data using 825 early-stage LUAD patients from 5 cohorts. Ranger algorithm was used to screen prognosis-associated biomarkers, which were confirmed with a validation phase. Clinical and biomarker information was fused using an iCluster plus algorithm, which significantly distinguished patients into high- and low-mortality risk groups ( = 0.01 and = 2.71×10). Further, potential functional DNA methylation-gene expression-overall survival pathways were evaluated by causal mediation analysis. The effect of DNA methylation level on LUAD survival was significantly mediated through gene expression level. By adding DNA methylation and gene expression biomarkers to a model of only clinical data, the AUCs of the trans-omics model improved by 18.3% (to 87.2%) and 16.4% (to 85.3%) in discovery and validation phases, respectively. Further, concordance index of the nomogram was 0.81 and 0.77 in discovery and validation phases, respectively. Based on systematic review of published literatures, our model was superior to all existing models for early-stage LUAD. In summary, our trans-omics model may help physicians accurately identify patients with high mortality risk.
有限的研究聚焦于利用跨组学生物标志物开发早期肺腺癌(LUAD)的预后模型。我们使用来自5个队列的825例早期LUAD患者,对临床信息、DNA甲基化和基因表达数据进行了综合分析。使用Ranger算法筛选与预后相关的生物标志物,并在验证阶段进行确认。使用iCluster plus算法融合临床和生物标志物信息,该算法显著将患者分为高死亡风险组和低死亡风险组( = 0.01, = 2.71×10)。此外,通过因果中介分析评估潜在的功能性DNA甲基化-基因表达-总生存途径。DNA甲基化水平对LUAD生存的影响通过基因表达水平显著介导。通过将DNA甲基化和基因表达生物标志物添加到仅包含临床数据的模型中,跨组学模型在发现阶段和验证阶段的AUC分别提高了18.3%(至87.2%)和16.4%(至85.3%)。此外,列线图在发现阶段和验证阶段的一致性指数分别为0.81和0.77。基于对已发表文献的系统综述,我们的模型优于所有现有的早期LUAD模型。总之,我们的跨组学模型可能有助于医生准确识别高死亡风险患者。