Bijvoet Center for Biomolecular Research, Faculty of Science - Chemistry, Utrecht University, Utrecht, the Netherlands.
Proteins. 2020 Feb;88(2):292-306. doi: 10.1002/prot.25802. Epub 2019 Sep 3.
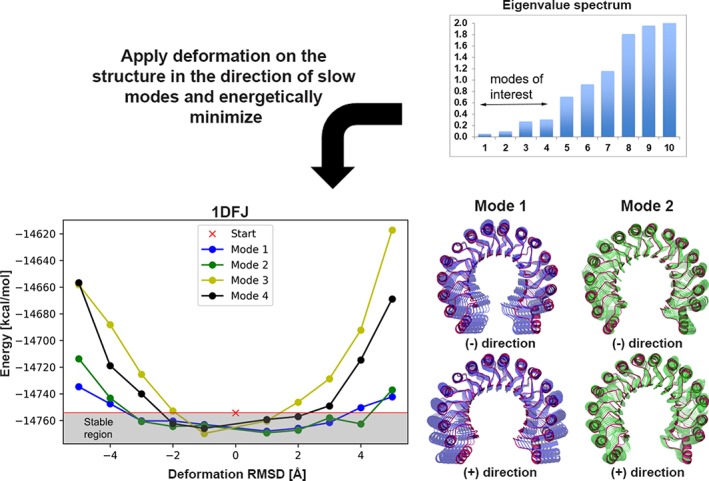
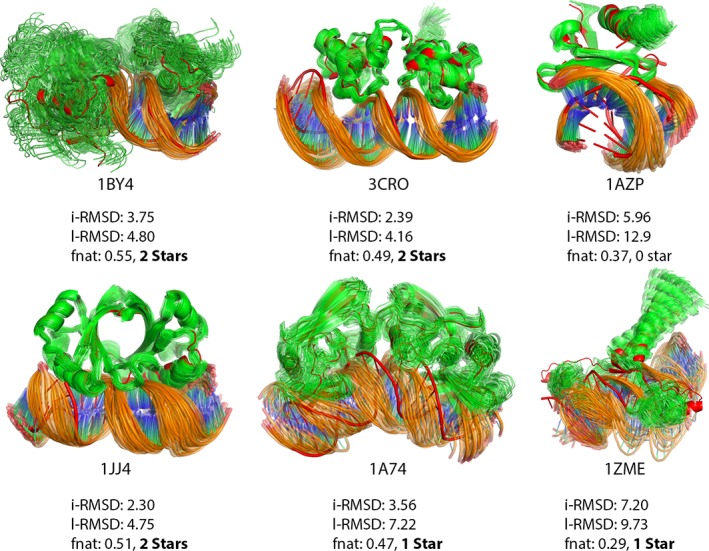
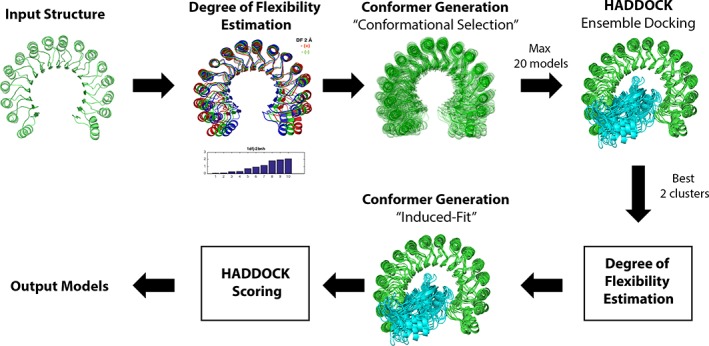
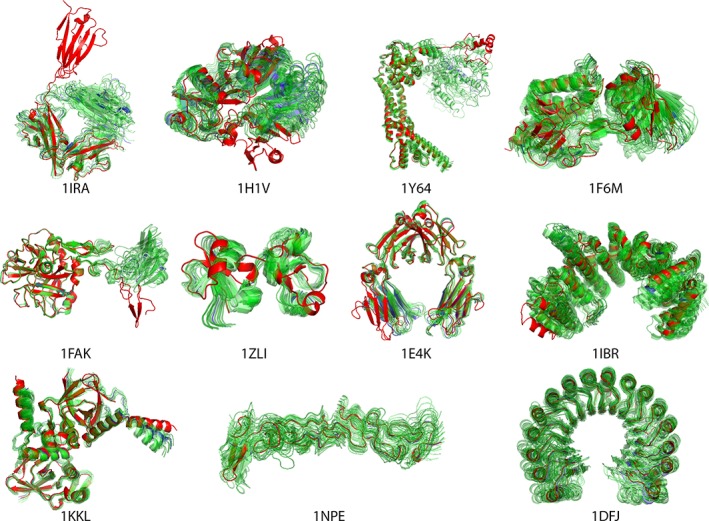
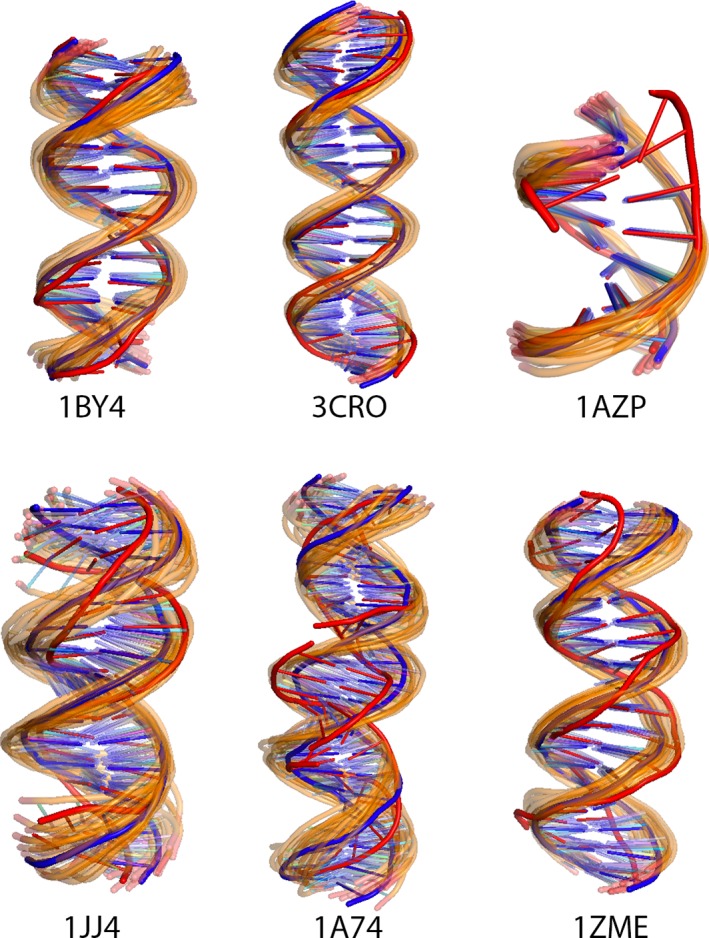
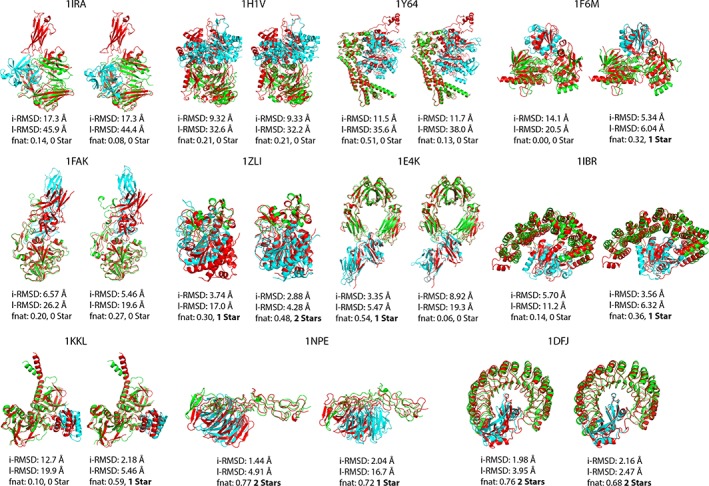
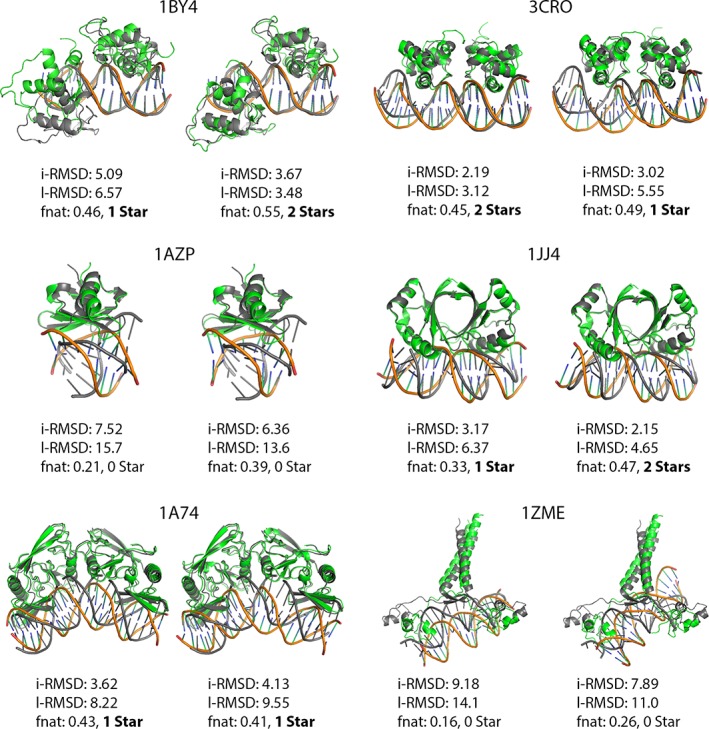
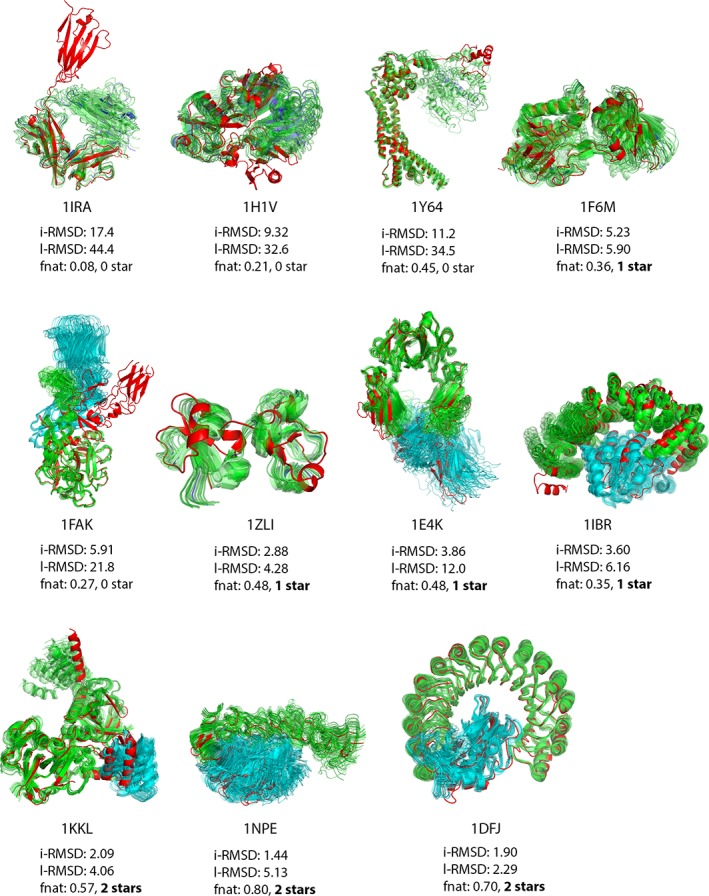
Incorporating the dynamic nature of biomolecules in the modeling of their complexes is a challenge, especially when the extent and direction of the conformational changes taking place upon binding is unknown. Estimating whether the binding of a biomolecule to its partner(s) occurs in a conformational state accessible to its unbound form ("conformational selection") and/or the binding process induces conformational changes ("induced-fit") is another challenge. We propose here a method combining conformational sampling using ClustENM-an elastic network-based modeling procedure-with docking using HADDOCK, in a framework that incorporates conformational selection and induced-fit effects upon binding. The extent of the applied deformation is estimated from its energetical costs, inspired from mechanical tensile testing on materials. We applied our pre- and post-docking sampling of conformational changes to the flexible multidomain protein-protein docking benchmark and a subset of the protein-DNA docking benchmark. Our ClustENM-HADDOCK approach produced acceptable to medium quality models in 7/11 and 5/6 cases for the protein-protein and protein-DNA complexes, respectively. The conformational selection (sampling prior to docking) has the highest impact on the quality of the docked models for the protein-protein complexes. The induced-fit stage of the pipeline (post-sampling), however, improved the quality of the final models for the protein-DNA complexes. Compared to previously described strategies to handle conformational changes, ClustENM-HADDOCK performs better than two-body docking in protein-protein cases but worse than a flexible multidomain docking approach. However, it does show a better or similar performance compared to previous protein-DNA docking approaches, which makes it a suitable alternative.
在对其复合物进行建模时纳入生物分子的动态性质是一个挑战,特别是当结合时发生的构象变化的程度和方向未知时。估计生物分子与其伴侣(多个)结合是否发生在其未结合形式可及的构象状态中(“构象选择”)和/或结合过程诱导构象变化(“诱导契合”)是另一个挑战。我们在这里提出了一种方法,该方法结合了使用 ClustENM(基于弹性网络的建模过程)进行构象采样和使用 HADDOCK 对接,该框架结合了结合时的构象选择和诱导契合效应。应用的变形程度是从其能量成本估计的,灵感来自对材料的机械拉伸测试。我们将预对接和对接后构象变化的采样应用于灵活的多域蛋白-蛋白对接基准测试和蛋白-DNA 对接基准测试的子集。我们的 ClustENM-HADDOCK 方法分别在 7/11 和 5/6 种蛋白-蛋白和蛋白-DNA 复合物中产生了可接受至中等质量的模型。对于蛋白-蛋白复合物,构象选择(对接前采样)对对接模型的质量影响最大。然而,该管道的诱导契合阶段(后采样)提高了最终蛋白-DNA 复合物模型的质量。与以前描述的处理构象变化的策略相比,ClustENM-HADDOCK 在蛋白-蛋白情况下的性能优于二体对接,但不如灵活的多域对接方法。然而,与以前的蛋白-DNA 对接方法相比,它表现出更好或相似的性能,使其成为一种合适的替代方法。