Science for Life Laboratory, 172 21, Solna, Sweden.
Department of Biochemistry and Biophysics, Stockholm University, 106 91, Stockholm, Sweden.
Nat Commun. 2022 Mar 10;13(1):1265. doi: 10.1038/s41467-022-28865-w.
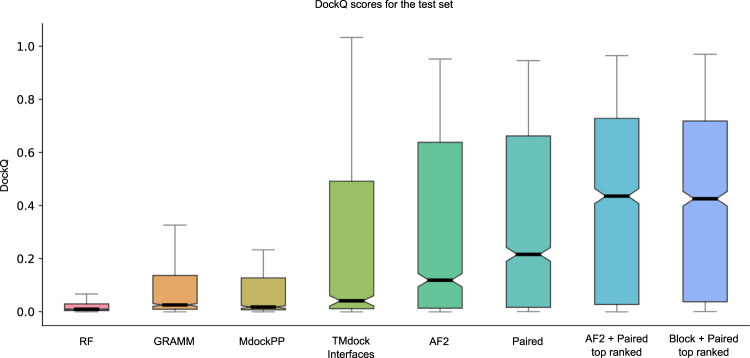
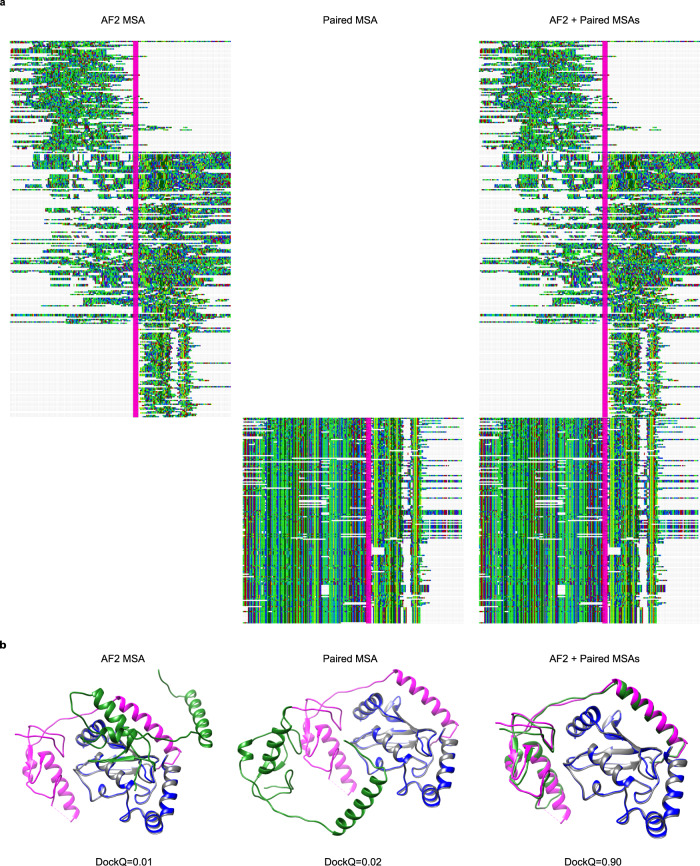
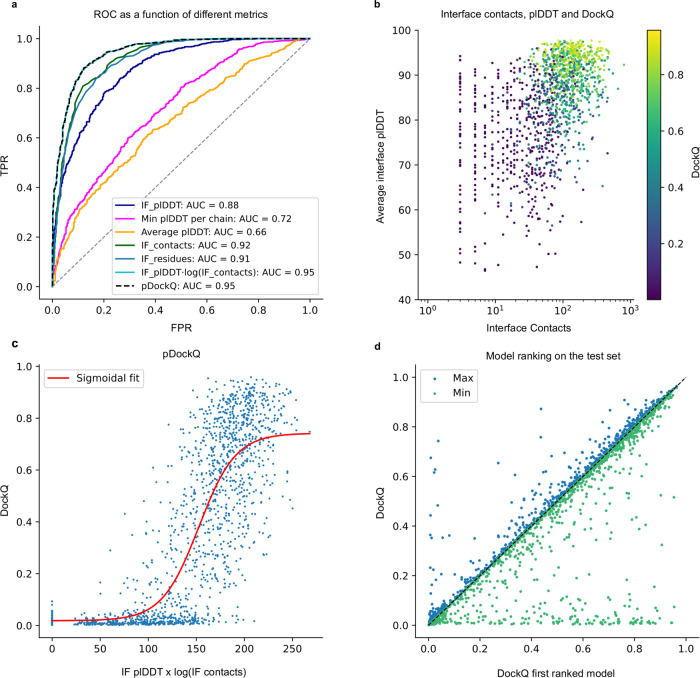
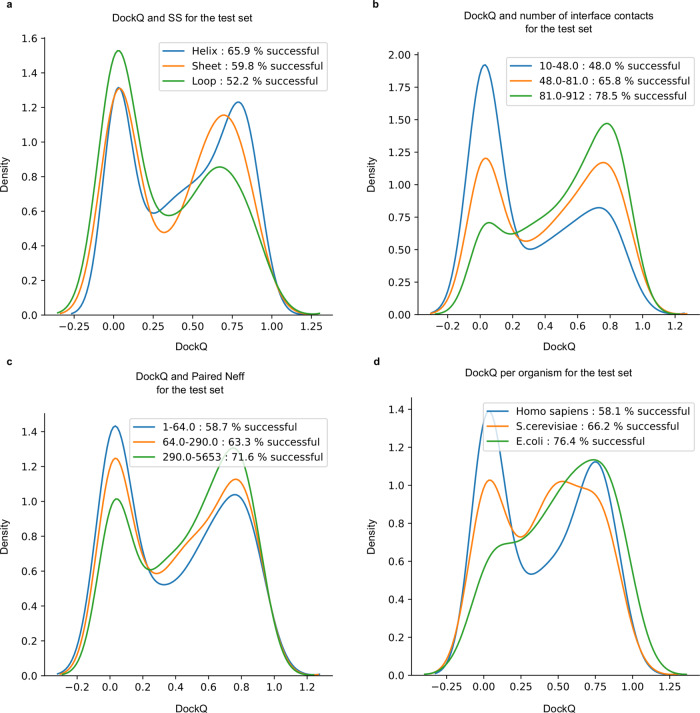
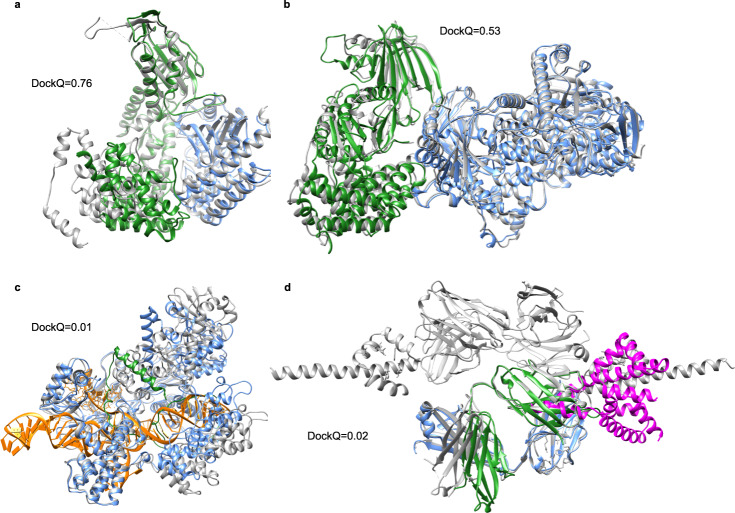
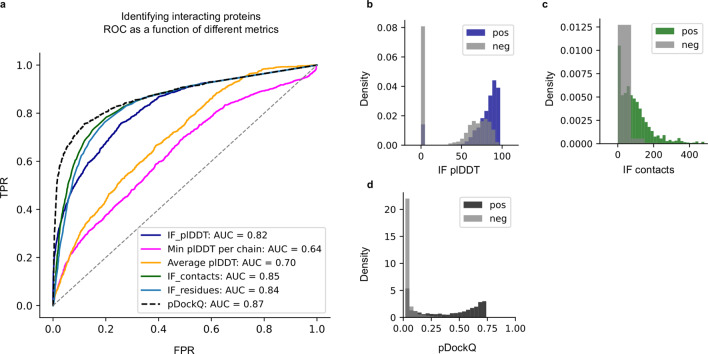
Predicting the structure of interacting protein chains is a fundamental step towards understanding protein function. Unfortunately, no computational method can produce accurate structures of protein complexes. AlphaFold2, has shown unprecedented levels of accuracy in modelling single chain protein structures. Here, we apply AlphaFold2 for the prediction of heterodimeric protein complexes. We find that the AlphaFold2 protocol together with optimised multiple sequence alignments, generate models with acceptable quality (DockQ ≥ 0.23) for 63% of the dimers. From the predicted interfaces we create a simple function to predict the DockQ score which distinguishes acceptable from incorrect models as well as interacting from non-interacting proteins with state-of-art accuracy. We find that, using the predicted DockQ scores, we can identify 51% of all interacting pairs at 1% FPR.
预测相互作用的蛋白质链的结构是理解蛋白质功能的基本步骤。不幸的是,没有计算方法可以生成蛋白质复合物的准确结构。AlphaFold2 在建模单链蛋白质结构方面表现出了前所未有的准确性。在这里,我们将 AlphaFold2 应用于预测异源二聚体蛋白质复合物。我们发现,AlphaFold2 方案加上优化的多重序列比对,可以为 63%的二聚体生成具有可接受质量(DockQ≥0.23)的模型。从预测的界面中,我们创建了一个简单的函数来预测 DockQ 分数,该分数可以区分可接受的和不正确的模型,以及具有最先进准确性的相互作用和非相互作用的蛋白质。我们发现,使用预测的 DockQ 分数,我们可以在 1%的 FPR 下识别所有相互作用对的 51%。