Department of Molecular Biology & Biochemistry and the Institute for Immunology, University of California, Irvine, California, United States of America.
PLoS Pathog. 2019 Aug 26;15(8):e1007923. doi: 10.1371/journal.ppat.1007923. eCollection 2019 Aug.
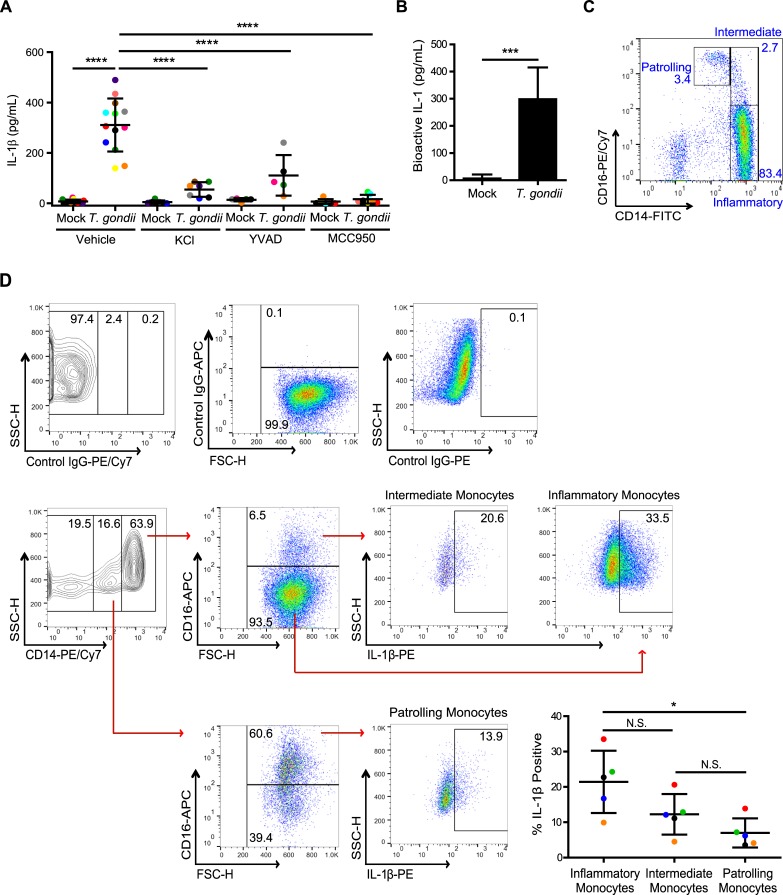
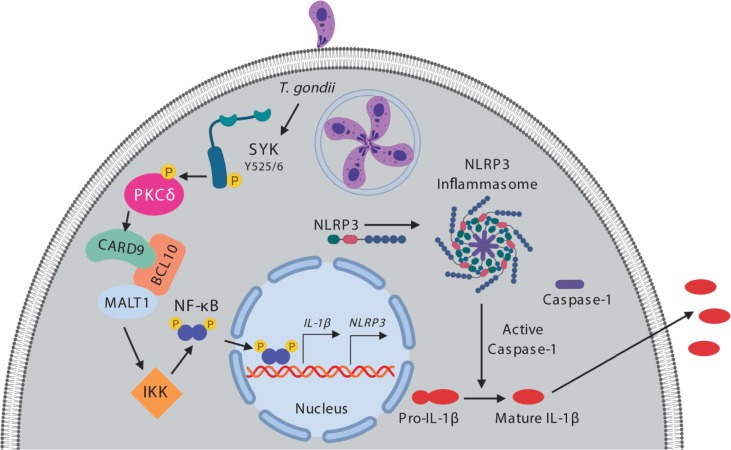
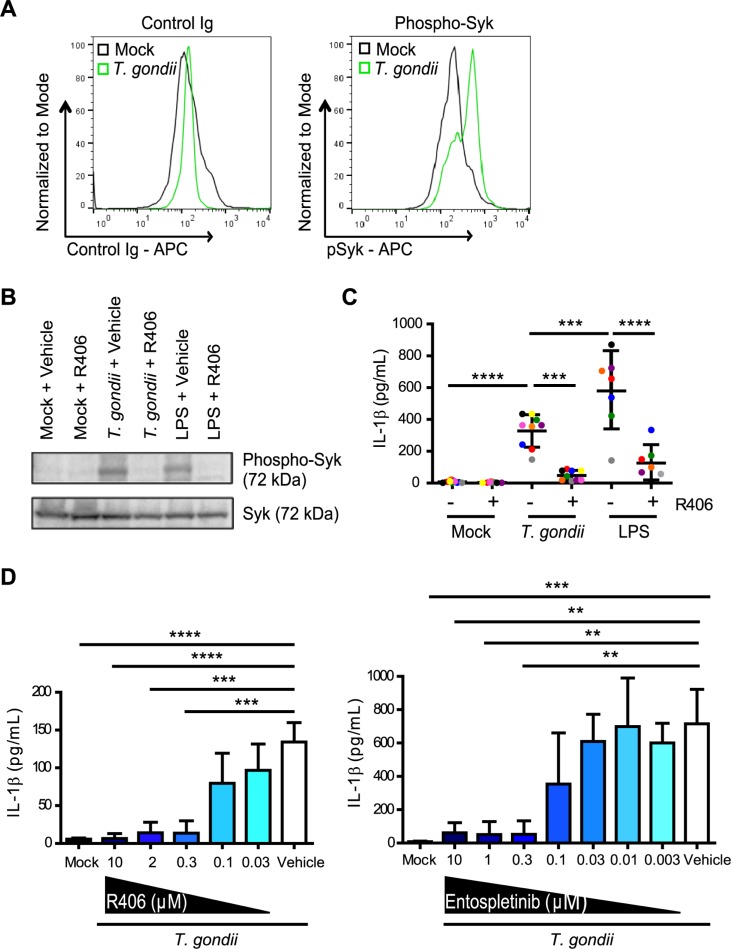
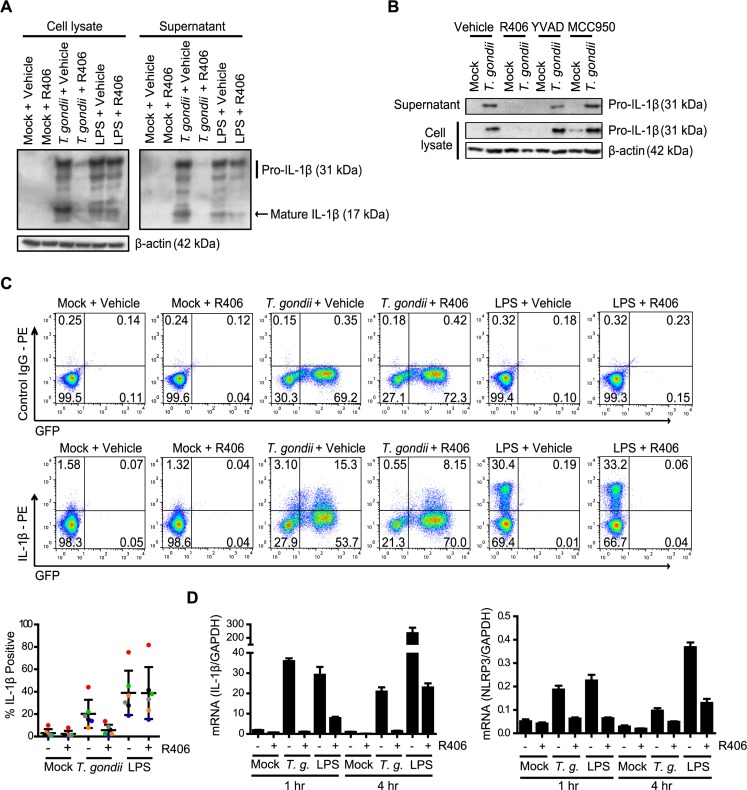
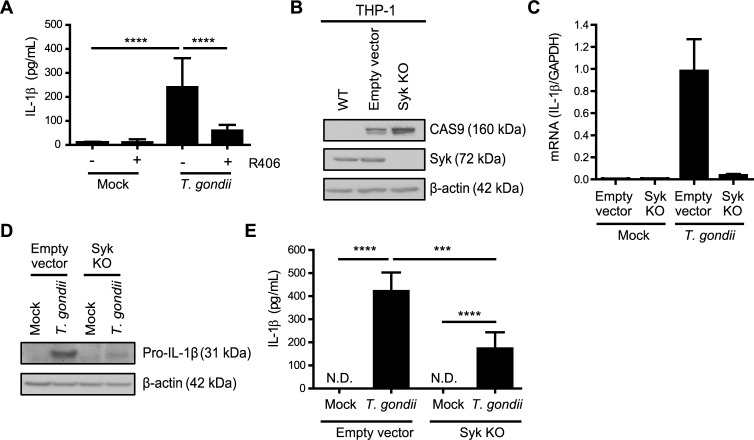
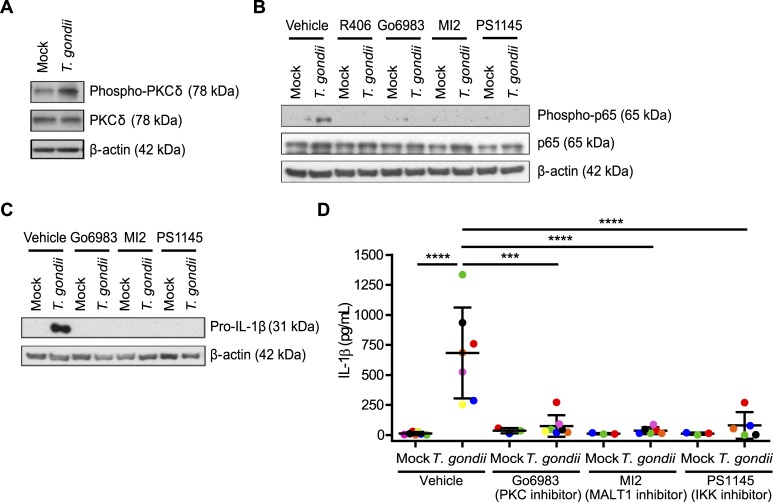
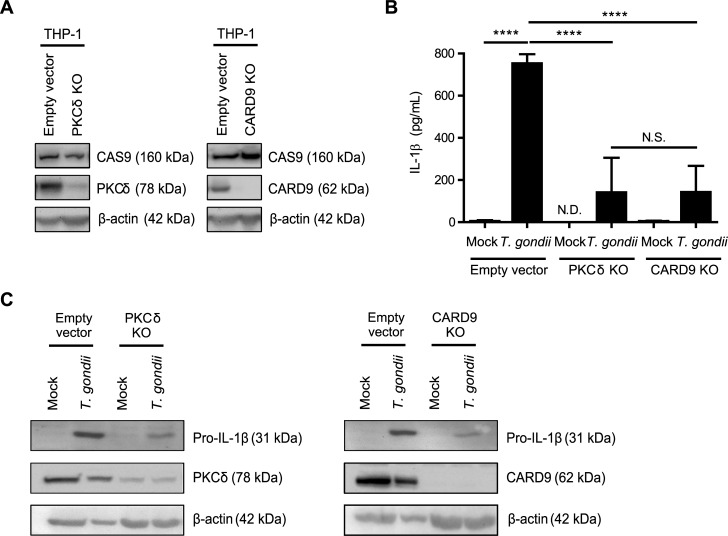
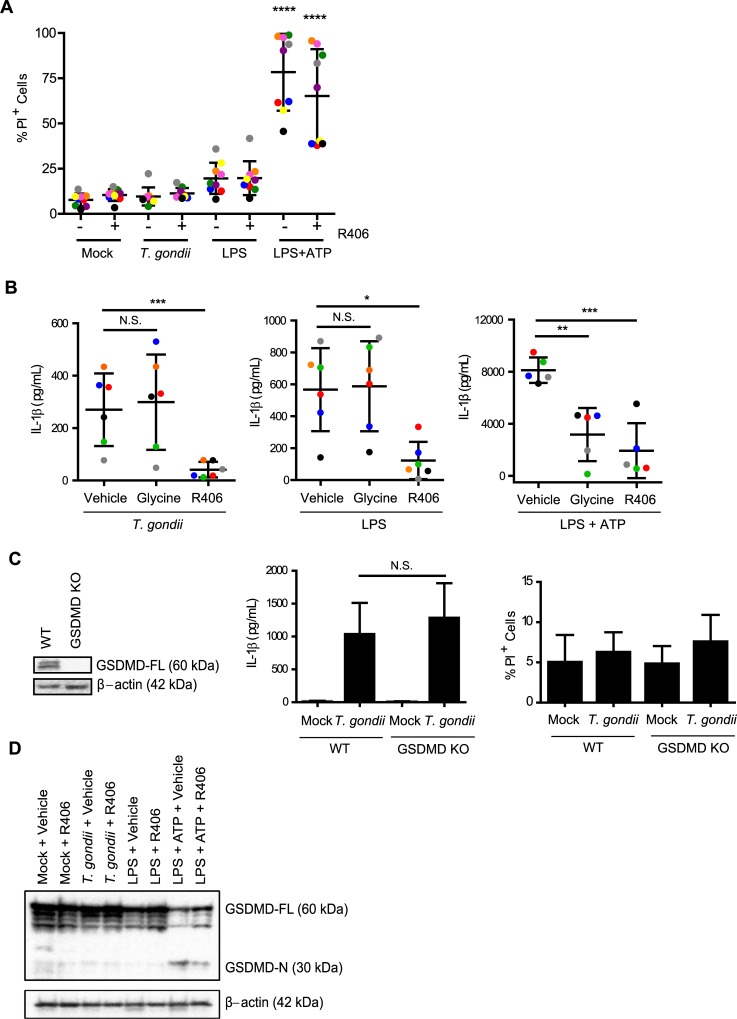
IL-1β is a potent pro-inflammatory cytokine that promotes immunity and host defense, and its dysregulation is associated with immune pathology. Toxoplasma gondii infection of myeloid cells triggers the production and release of IL-1β; however, the mechanisms regulating this pathway, particularly in human immune cells, are incompletely understood. We have identified a novel pathway of T. gondii induction of IL-1β via a Syk-CARD9-NF-κB signaling axis in primary human peripheral blood monocytes. Syk was rapidly phosphorylated during T. gondii infection of primary monocytes, and inhibiting Syk with the pharmacological inhibitors R406 or entospletinib, or genetic ablation of Syk in THP-1 cells, reduced IL-1β release. Inhibition of Syk in primary cells or deletion of Syk in THP-1 cells decreased parasite-induced IL-1β transcripts and the production of pro-IL-1β. Furthermore, inhibition of PKCδ, CARD9/MALT-1 and IKK reduced p65 phosphorylation and pro-IL-1β production in T. gondii-infected primary monocytes, and genetic knockout of PKCδ or CARD9 in THP-1 cells also reduced pro-IL-1β protein levels and IL-1β release during T. gondii infection, indicating that Syk functions upstream of this NF-κB-dependent signaling pathway for IL-1β transcriptional activation. IL-1β release from T. gondii-infected primary human monocytes required the NLRP3-caspase-1 inflammasome, but interestingly, was independent of gasdermin D (GSDMD) cleavage and pyroptosis. Moreover, GSDMD knockout THP-1 cells released comparable amounts of IL-1β to wild-type THP-1 cells after T. gondii infection. Taken together, our data indicate that T. gondii induces a Syk-CARD9/MALT-1-NF-κB signaling pathway and activation of the NLRP3 inflammasome for the release of IL-1β in a cell death- and GSDMD-independent manner. This research expands our understanding of the molecular basis for human innate immune regulation of inflammation and host defense during parasite infection.
白细胞介素-1β(IL-1β)是一种有效的促炎细胞因子,可促进免疫和宿主防御,其失调与免疫病理学有关。刚地弓形虫感染髓系细胞会触发 IL-1β的产生和释放;然而,调节该途径的机制,特别是在人类免疫细胞中,尚不完全清楚。我们已经确定了一种新的途径,即通过 Syk-CARD9-NF-κB 信号轴诱导人外周血单核细胞产生 IL-1β。在原代单核细胞感染刚地弓形虫期间,Syk 迅速磷酸化,用药理学抑制剂 R406 或 entospletinib 抑制 Syk,或在 THP-1 细胞中基因敲除 Syk,均可减少 IL-1β的释放。在原代细胞中抑制 Syk 或在 THP-1 细胞中敲除 Syk 可减少寄生虫诱导的 IL-1β 转录物和 pro-IL-1β 的产生。此外,抑制 PKCδ、CARD9/MALT-1 和 IKK 可减少感染刚地弓形虫的原代单核细胞中 p65 的磷酸化和 pro-IL-1β 的产生,在 THP-1 细胞中基因敲除 PKCδ 或 CARD9 也可减少 pro-IL-1β 蛋白水平和刚地弓形虫感染期间的 IL-1β 释放,表明 Syk 在该 NF-κB 依赖的 IL-1β 转录激活信号通路中起上游作用。刚地弓形虫感染的原代人单核细胞释放的 IL-1β 需要 NLRP3-caspase-1 炎性体,但有趣的是,不需要 gasdermin D(GSDMD)裂解和细胞焦亡。此外,刚地弓形虫感染后,GSDMD 敲除的 THP-1 细胞释放的 IL-1β 与野生型 THP-1 细胞相当。综上所述,我们的数据表明,刚地弓形虫诱导 Syk-CARD9/MALT-1-NF-κB 信号通路和 NLRP3 炎性体的激活,以非细胞死亡和 GSDMD 依赖的方式释放 IL-1β。这项研究扩展了我们对寄生虫感染期间人类先天免疫调节炎症和宿主防御的分子基础的理解。