Guo Yonglong, Wang Peiyuan, Ma Jacey Hongjie, Cui Zekai, Yu Quan, Liu Shiwei, Xue Yunxia, Zhu Deliang, Cao Jixing, Li Zhijie, Tang Shibo, Chen Jiansu
Ophthalmology Department, The First Affiliated Hospital of Jinan University, Guangzhou, China.
Key Laboratory for Regenerative Medicine of Ministry of Education, Jinan University, Guangzhou, China.
Front Cell Neurosci. 2019 Aug 7;13:361. doi: 10.3389/fncel.2019.00361. eCollection 2019.
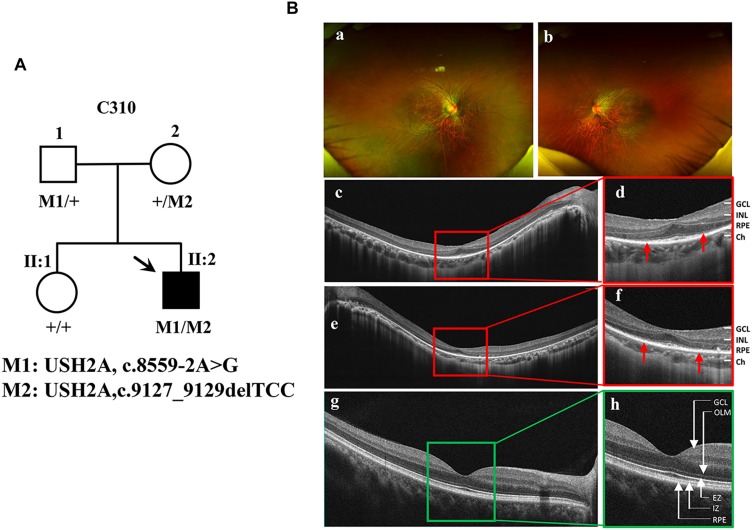
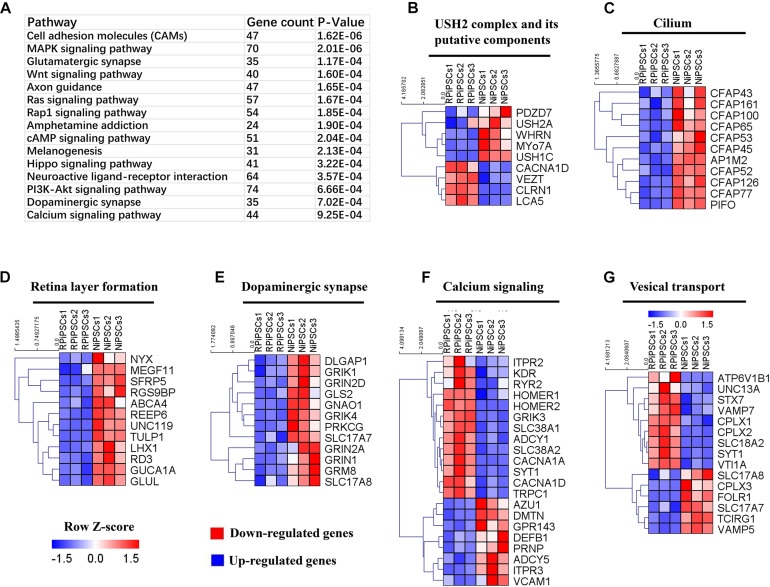
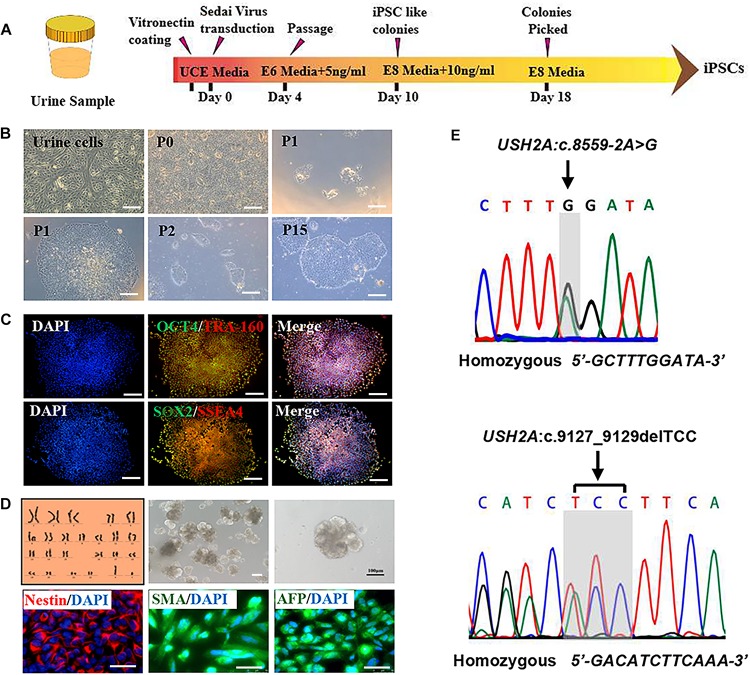
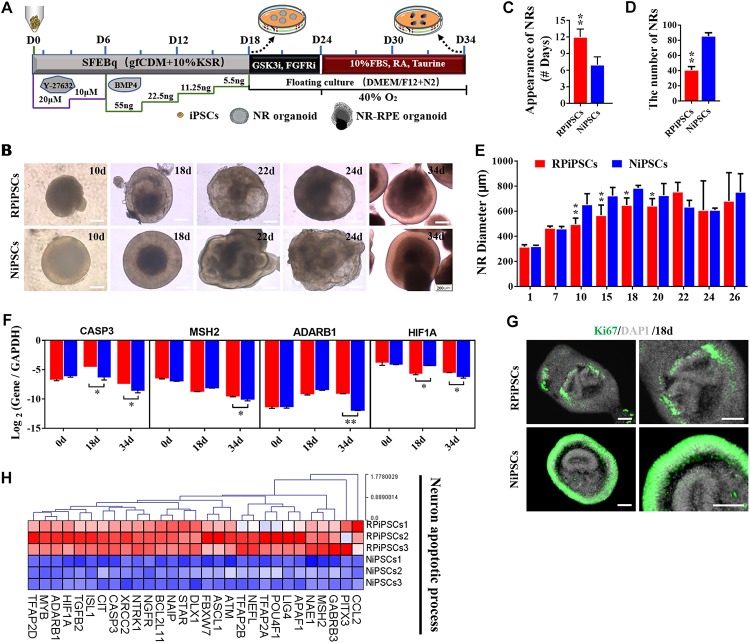
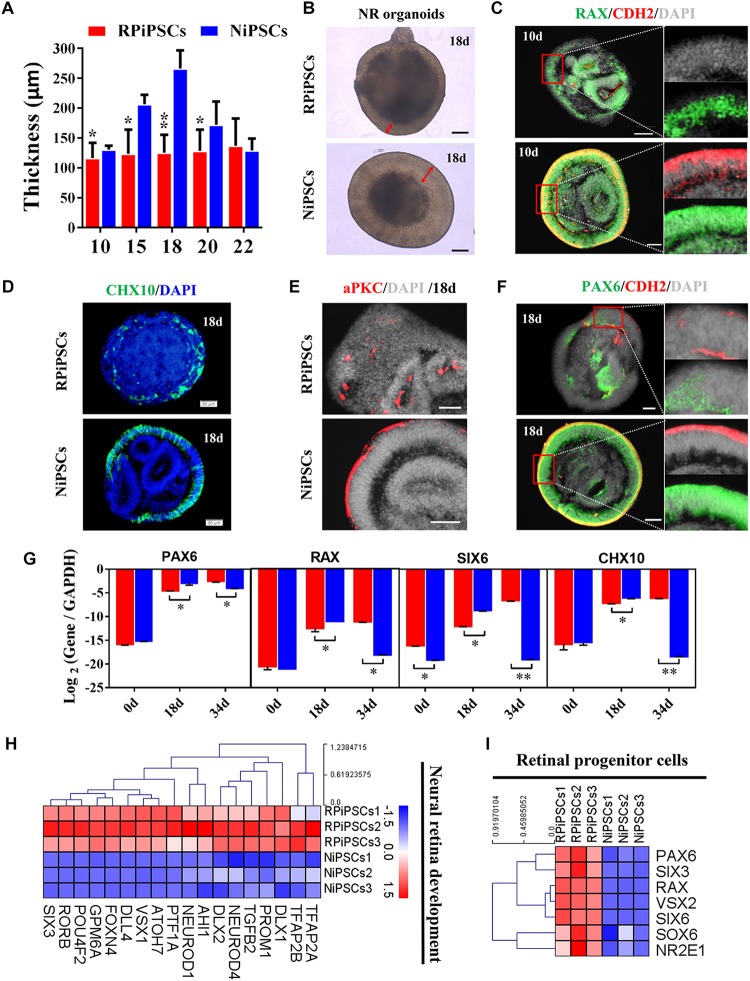
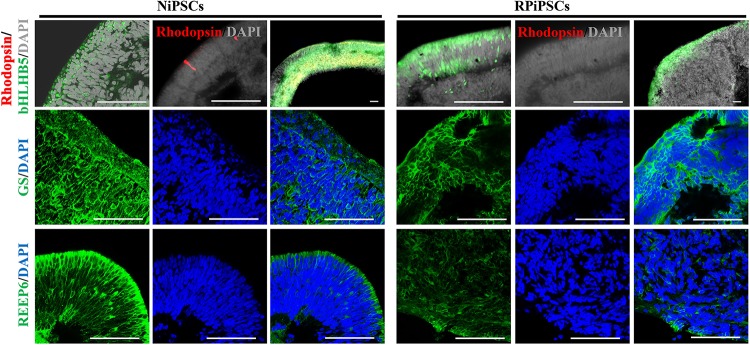
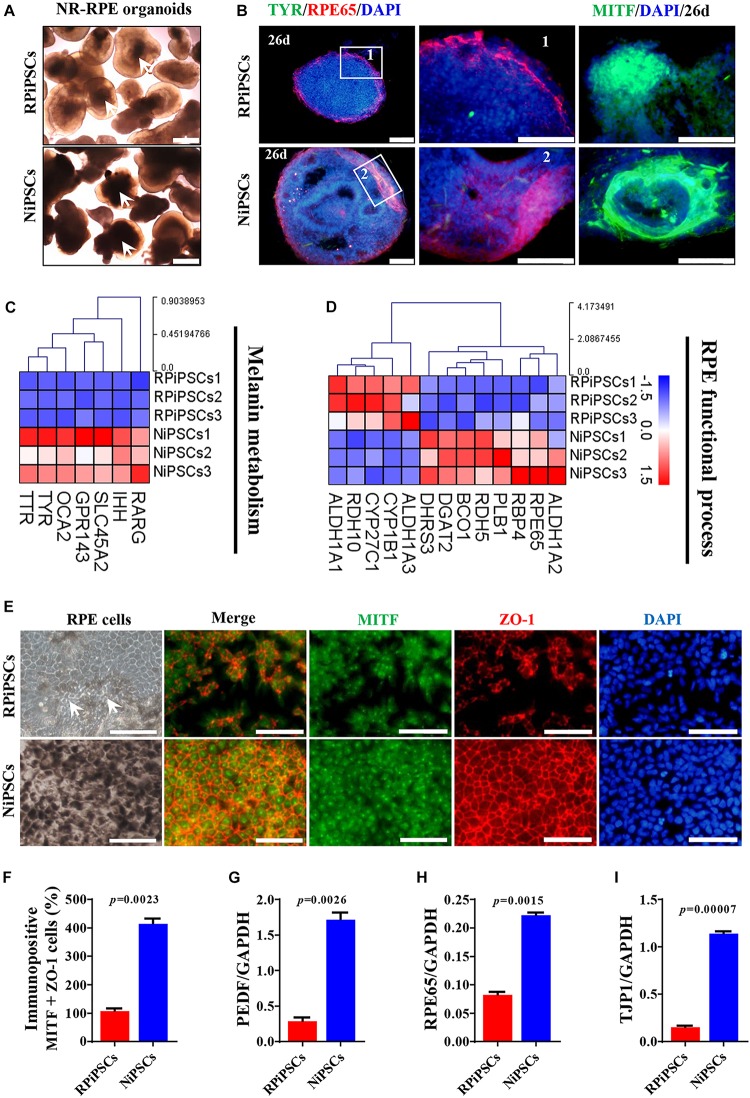
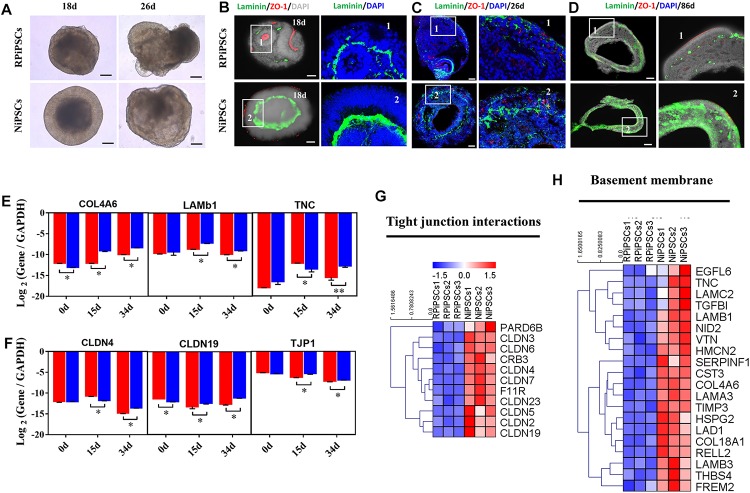
Retinitis pigmentosa (RP) represents a group of inherited retinopathies with early-onset nyctalopia followed by progressive photoreceptor degeneration causing irreversible vision loss. Mutations in USH2A are the most common cause of non-syndromic RP. Here, we reprogrammed induced pluripotent stem cells (iPSCs) from a RP patient with a mutation in (c.8559-2A > G/c.9127_9129delTCC). Then, multilayer retinal organoids including neural retina (NR) and retinal pigment epithelium (RPE) were generated by three-step "induction-reversal culture." The early retinal organoids derived from the RP patient with the USH2A mutation exhibited significant defects in terms of morphology, immunofluorescence staining and transcriptional profiling. To the best of our knowledge, the pathogenic mutation (c.9127_9129delTCC) in has not been reported previously among RP patients. Notably, the expression of laminin in the USH2A mutation organoids was significantly lower than in the iPSCs derived from healthy, age- and sex-matched controls during the retinal organogenesis. We also observed that abnormal retinal neuroepithelium differentiation and polarization caused defective retinal progenitor cell development and retinal layer formation, disordered organization of NRs in the presence of the USH2A mutation. Furthermore, the USH2A mutation bearing RPE cells presented abnormal morphology, lacking pigmented foci and showing an apoptotic trend and reduced expression of specific makers, such as MITF, , and RPE65. In addition, the USH2A mutation organoids had lower expression of cilium-associated (especially , ) and dopaminergic synapse-related genes (including , , , and ), while there was higher expression of neuron apoptotic process-related genes (especially , , and ). This study may provide essential assistance in the molecular diagnosis and screening of RP. This work recapitulates the pathogenesis of USH2A using patient-specific organoids and demonstrated that alterations in USH2A function due to mutations may lead to cellular and molecular abnormalities.
视网膜色素变性(RP)是一组遗传性视网膜病变,其早期症状为夜盲症,随后是进行性光感受器退化,导致不可逆转的视力丧失。USH2A基因的突变是导致非综合征性RP的最常见原因。在此,我们对一名患有(c.8559-2A>G/c.9127_9129delTCC)突变的RP患者的诱导多能干细胞(iPSC)进行了重编程。然后,通过三步“诱导-逆转培养”生成了包括神经视网膜(NR)和视网膜色素上皮(RPE)的多层视网膜类器官。来自患有USH2A突变的RP患者的早期视网膜类器官在形态、免疫荧光染色和转录谱方面表现出明显缺陷。据我们所知,此前在RP患者中尚未报道过(c.9127_9129delTCC)这一致病突变。值得注意的是,在视网膜器官发生过程中,USH2A突变类器官中层粘连蛋白的表达明显低于来自健康、年龄和性别匹配对照的iPSC。我们还观察到,异常的视网膜神经上皮分化和极化导致视网膜祖细胞发育和视网膜层形成缺陷,在存在USH2A突变的情况下NRs组织紊乱。此外,携带USH2A突变的RPE细胞呈现出异常形态,缺乏色素沉着灶,呈现凋亡趋势,且特定标志物如MITF、 和RPE65的表达降低。此外,USH2A突变类器官中与纤毛相关(尤其是 、 )和多巴胺能突触相关基因(包括 、 、 、和 )的表达较低,而与神经元凋亡过程相关基因(尤其是 、 、和 )的表达较高。本研究可能为RP的分子诊断和筛查提供重要帮助。这项工作利用患者特异性类器官概括了USH2A的发病机制,并证明由于突变导致的USH2A功能改变可能导致细胞和分子异常。