Nuffield Department of Clinical Neurosciences, John Radcliffe Hospital, University of Oxford, Oxford, United Kingdom.
Perelman Center for Advanced Medicine, Department of Neurology, University of Pennsylvania, Philadelphia.
JAMA Neurol. 2020 Jan 1;77(1):82-93. doi: 10.1001/jamaneurol.2019.2940.
Identifying the course of demyelinating disease associated with myelin oligodendrocyte glycoprotein (MOG) autoantibodies is critical to guide appropriate treatment choices.
To characterize serial anti-MOG antibody serologies and clinical and imaging features at presentation and during follow-up in an inception cohort of prospectively monitored children with acquired demyelination.
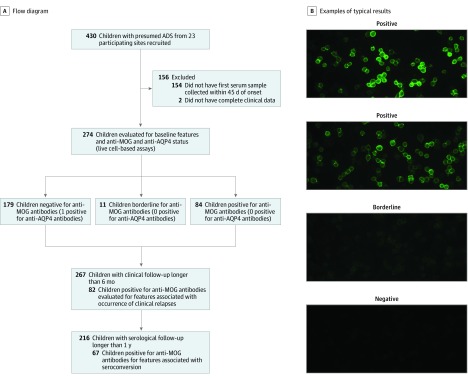
DESIGN, SETTING, AND PARTICIPANTS: In this prospective cohort study, study participants were recruited from July 2004 to February 2017 through the multicenter Canadian Pediatric Demyelinating Disease Study. Inclusion criteria included (1) incident central nervous system demyelination, (2) at least 1 serum sample obtained within 45 days from onset, and (3) complete clinical information. Of 430 participants with acquired demyelinating syndrome recruited, 274 were included in analyses. Of 156 excluded participants, 154 were excluded owing to missing baseline samples and 2 owing to incomplete clinical information. Data were analyzed from May to October 2018.
Presence of anti-MOG antibodies was blindly assessed in serial samples collected over a median of 4 years. Clinical, magnetic resonance imaging, and cerebrospinal fluid features were characterized at presentation, and subsequent disease course was assessed by development of new brain magnetic resonance imaging lesions, total lesion volume at last evaluation, annualized relapse rates, Expanded Disability Status Scale score and visual functional score at 4 years, and any disease-modifying treatment exposure.
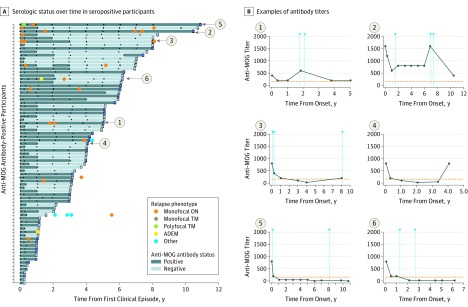
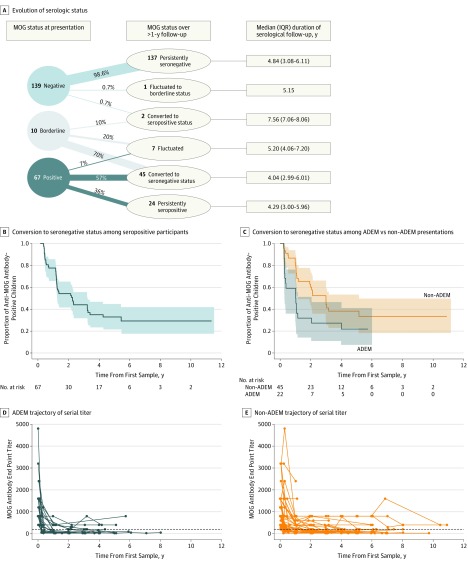
Of the 274 included participants, 140 (51.1%) were female, and the median (interquartile range) age of all participants was 10.8 (6.2-13.9) years. One-third of children were positive for anti-MOG antibodies at the time of incident demyelination. Clinical presentations included a combination of optic neuritis, transverse myelitis, and acute disseminated encephalomyelitis for 81 of 84 anti-MOG antibody-positive children (96%). Brain lesions were present in 51 of 76 anti-MOG antibody-positive participants (67%), but magnetic resonance imaging characteristics differed with age at presentation. Complete resolution of baseline lesions was observed in 26 of 49 anti-MOG antibody-positive participants (53%). On serial serum analysis, 38 of 67 participants (57%) who were seropositive at onset became seronegative (median time to conversion, 1 year). Among all participants who were positive for anti-MOG antibodies at presentation, clinical relapses occurred in 9 of 24 children (38%) who remained persistently seropositive and in 5 of 38 children (13%) who converted to seronegative status.
Myelin oligodendrocyte glycoprotein antibodies are common in children with acquired demyelinating syndrome and are transient in approximatively half of cases. Even when persistently positive, most anti-MOG antibody-positive children experience a monophasic disease. The presence of anti-MOG antibodies at the time of incident demyelination should not immediately prompt the initiation of long-term immunomodulatory therapy.
确定与髓鞘少突胶质细胞糖蛋白(MOG)自身抗体相关的脱髓鞘疾病进程对于指导适当的治疗选择至关重要。
在一个前瞻性监测的儿童获得性脱髓鞘疾病队列中,描述抗 MOG 抗体血清学和临床及影像学特征在发病时和随访期间的变化。
设计、地点和参与者:在这项前瞻性队列研究中,研究参与者于 2004 年 7 月至 2017 年 2 月通过加拿大儿科脱髓鞘疾病研究的多中心项目招募。纳入标准包括:(1)中枢神经系统脱髓鞘疾病首次发作,(2)至少有 1 份在发病后 45 天内采集的血清样本,(3)有完整的临床资料。在招募的 430 例获得性脱髓鞘综合征患者中,有 274 例被纳入分析。在 156 例排除的患者中,有 154 例因基线样本缺失而被排除,2 例因临床资料不完整而被排除。数据分析于 2018 年 5 月至 10 月进行。
在中位数为 4 年的时间内,对收集的系列样本进行了抗 MOG 抗体的盲法评估。在发病时对临床、磁共振成像和脑脊液特征进行了描述,并通过新的脑磁共振成像病灶的发展、最后评估时的总病灶体积、年复发率、4 年时的扩展残疾状态量表评分和视觉功能评分、以及任何疾病修正治疗的暴露情况来评估后续的疾病进程。
在纳入的 274 例患者中,140 例(51.1%)为女性,所有患者的中位(四分位距)年龄为 10.8(6.2-13.9)岁。三分之一的儿童在脱髓鞘发作时抗 MOG 抗体呈阳性。临床表现包括 84 例抗 MOG 抗体阳性儿童中的 81 例(96%)视神经炎、横贯性脊髓炎和急性播散性脑脊髓炎的组合。51 例(67%)抗 MOG 抗体阳性患者存在脑部病变,但磁共振成像特征随发病时的年龄而变化。49 例(53%)抗 MOG 抗体阳性患者基线病变完全消退。在连续的血清学分析中,在 67 例发病时血清阳性的患者中,有 38 例(57%)转为血清阴性(中位转换时间为 1 年)。在所有在发病时抗 MOG 抗体阳性的患者中,24 例(38%)持续血清阳性的患者和 38 例(13%)转为血清阴性的患者发生了临床复发。
髓鞘少突胶质细胞糖蛋白抗体在儿童获得性脱髓鞘综合征中很常见,约一半的病例为一过性。即使持续阳性,大多数抗 MOG 抗体阳性的儿童也经历单相疾病。在脱髓鞘发作时存在抗 MOG 抗体不应立即提示启动长期免疫调节治疗。