Ehsan Asma, Mahmood Muhammad K, Khan Yaser D, Barukab Omar M, Khan Sher A, Chou Kuo-Chen
1Department of Mathematics, University of the Punjab, Lahore, Pakistan; 2Faculty of Information Technology, University of Management and Technology, Lahore, Pakistan; 3King Abdul Aziz University, Faculty of Computing and Information Technology in Rabigh, Jeddah, KSA; 4Gordon Life Science Institute, Boston, MA 02478, USA.
Curr Genomics. 2019 Feb;20(2):124-133. doi: 10.2174/1389202920666190325162307.
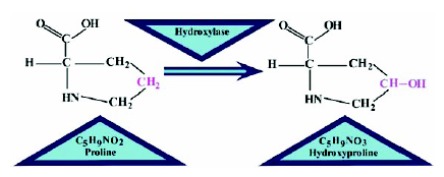
In various biological processes and cell functions, Post Translational Modifications (PTMs) bear critical significance. Hydroxylation of proline residue is one kind of PTM, which occurs following protein synthesis. The experimental determination of hydroxyproline sites in an uncharacterized protein sequence requires extensive, time-consuming and expensive tests.
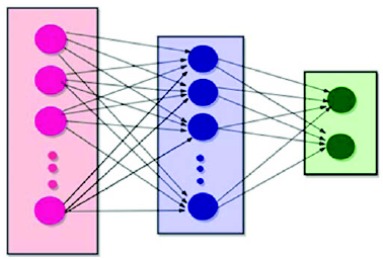
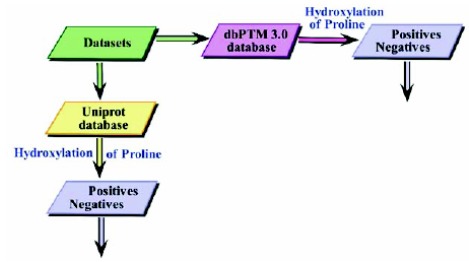
With the torrential slide of protein sequences produced in the post-genomic age, certain remarkable computational strategies are desired to overwhelm the issue. Keeping in view the composition and sequence order effect within polypeptide chains, an innovative predictor a mathematical model is proposed.
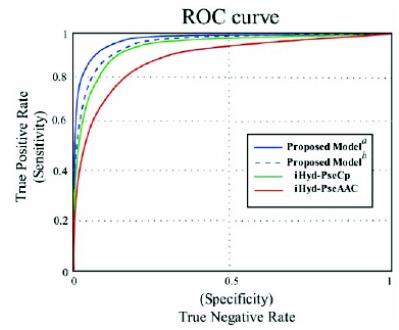
Later, it was stringently verified using self-consistency, cross-validation and jackknife tests on benchmark datasets. It was established after a rigorous jackknife test that the new predictor values are superior to the values predicted by previous methodologies.
This new mathematical technique is the most appropriate and encouraging as compared with the existing models.
在各种生物过程和细胞功能中,翻译后修饰(PTM)具有至关重要的意义。脯氨酸残基的羟基化是一种翻译后修饰,它在蛋白质合成后发生。在未表征的蛋白质序列中实验确定羟脯氨酸位点需要进行广泛、耗时且昂贵的测试。
随着后基因组时代产生的蛋白质序列大量涌现,需要某些显著的计算策略来解决这个问题。考虑到多肽链内的组成和序列顺序效应,提出了一种创新的预测器——一种数学模型。
随后,在基准数据集上使用自一致性、交叉验证和留一法测试对其进行了严格验证。经过严格的留一法测试后确定,新预测器的值优于先前方法预测的值。
与现有模型相比,这种新的数学技术是最合适且令人鼓舞的。