Department of Mathematics, School of Science, Xi'an Shiyou University, 710065 Xi'an, China.
School of Computer Science, Northwestern Polytechnical University, 710072 Xi'an, China.
Cells. 2019 Sep 27;8(10):1161. doi: 10.3390/cells8101161.
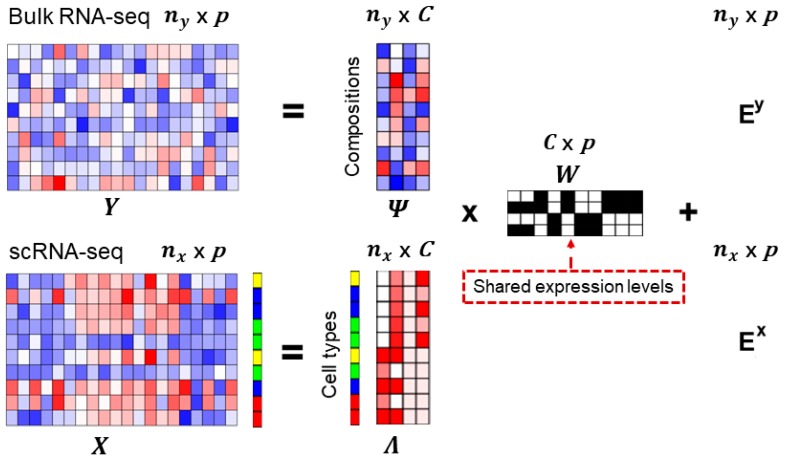
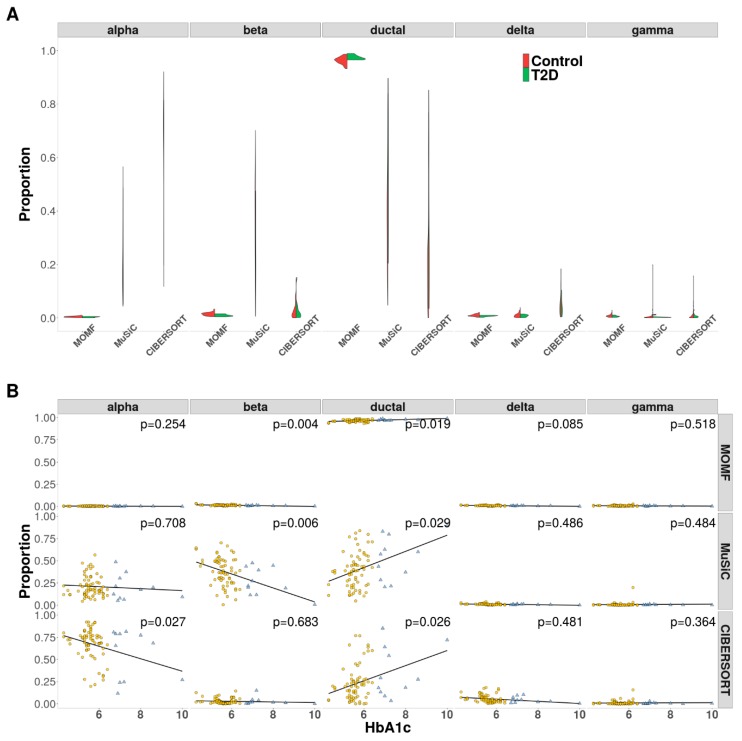
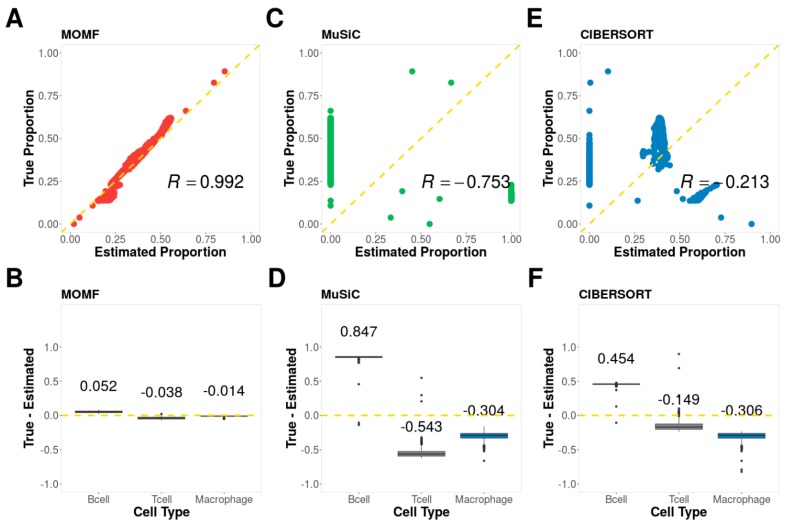
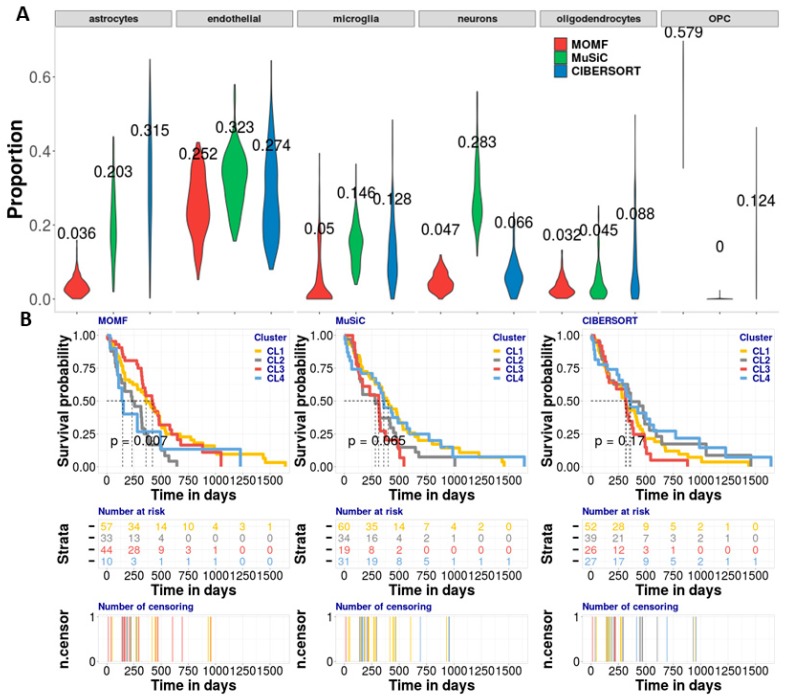
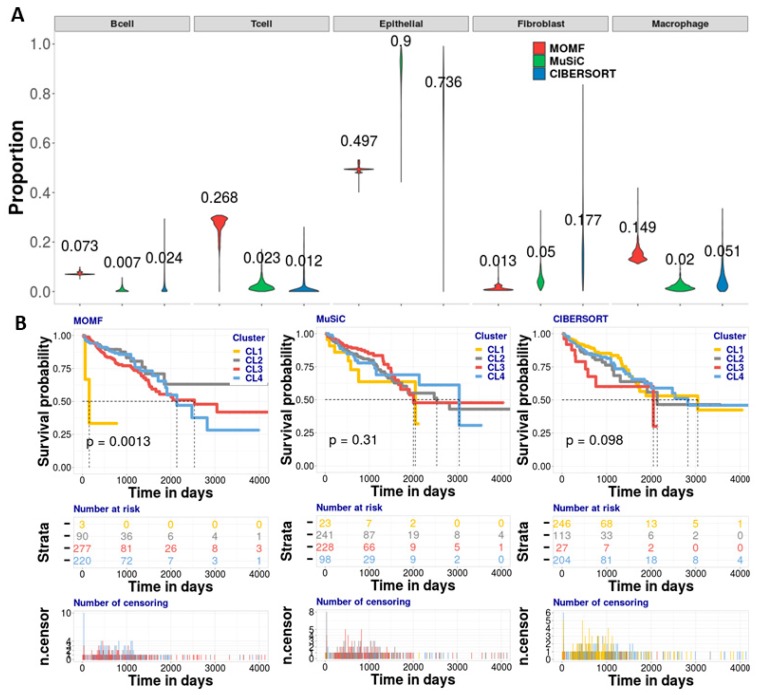
Estimating cell type compositions for complex diseases is an important step to investigate the cellular heterogeneity for understanding disease etiology and potentially facilitate early disease diagnosis and prevention. Here, we developed a computationally statistical method, referring to Multi-Omics Matrix Factorization (MOMF), to estimate the cell-type compositions of bulk RNA sequencing (RNA-seq) data by leveraging cell type-specific gene expression levels from single-cell RNA sequencing (scRNA-seq) data. MOMF not only directly models the count nature of gene expression data, but also effectively accounts for the uncertainty of cell type-specific mean gene expression levels. We demonstrate the benefits of MOMF through three real data applications, i.e., Glioblastomas (GBM), colorectal cancer (CRC) and type II diabetes (T2D) studies. MOMF is able to accurately estimate disease-related cell type proportions, i.e., oligodendrocyte progenitor cells and macrophage cells, which are strongly associated with the survival of GBM and CRC, respectively.
估计复杂疾病的细胞类型组成是研究细胞异质性以了解疾病病因并可能有助于早期疾病诊断和预防的重要步骤。在这里,我们开发了一种计算统计方法,称为多组学矩阵分解(MOMF),通过利用单细胞 RNA 测序(scRNA-seq)数据中细胞类型特异性基因表达水平,从批量 RNA 测序(RNA-seq)数据中估计细胞类型组成。MOMF 不仅直接对基因表达数据的计数性质进行建模,而且还能有效地考虑细胞类型特异性平均基因表达水平的不确定性。我们通过三个真实数据应用,即脑胶质瘤(GBM)、结直肠癌(CRC)和 II 型糖尿病(T2D)研究,展示了 MOMF 的优势。MOMF 能够准确估计与疾病相关的细胞类型比例,即少突胶质细胞祖细胞和巨噬细胞,它们与 GBM 和 CRC 的存活率密切相关。