Department of Biology, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China.
World J Gastroenterol. 2019 Sep 28;25(36):5434-5450. doi: 10.3748/wjg.v25.i36.5434.
High mobility group box-1 (HMGB1), recognized as a representative of damage-associated molecular patterns, is released during cell injury/death, triggering the inflammatory response and ultimately resulting in tissue damage. Dozens of studies have shown that HMGB1 is involved in certain diseases, but the details on how injured hepatocytes release HMGB1 need to be elicited.
To reveal HMGB1 release mechanism in hepatocytes undergoing oxidative stress.
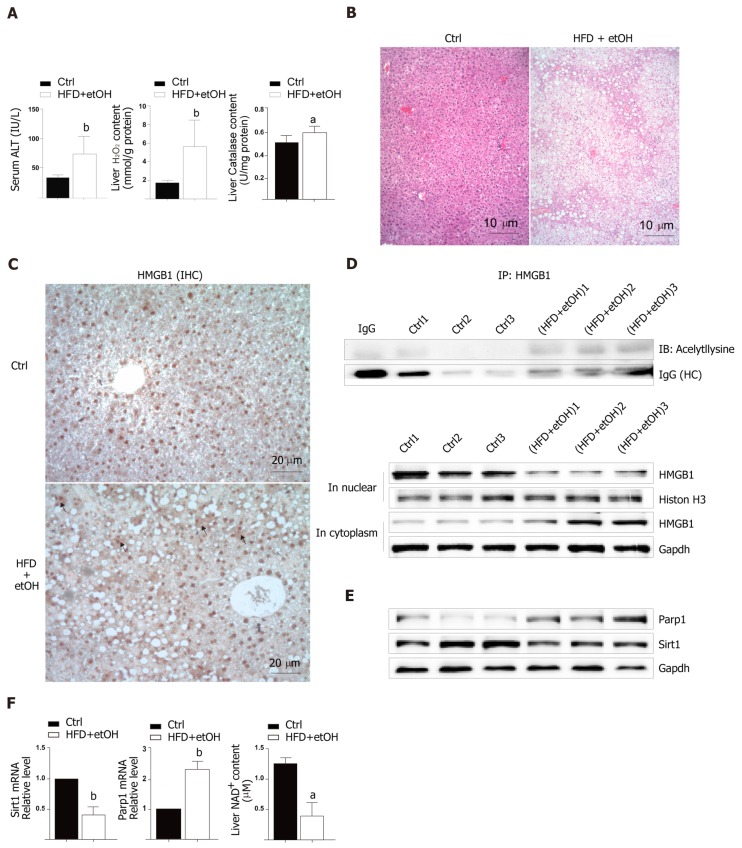
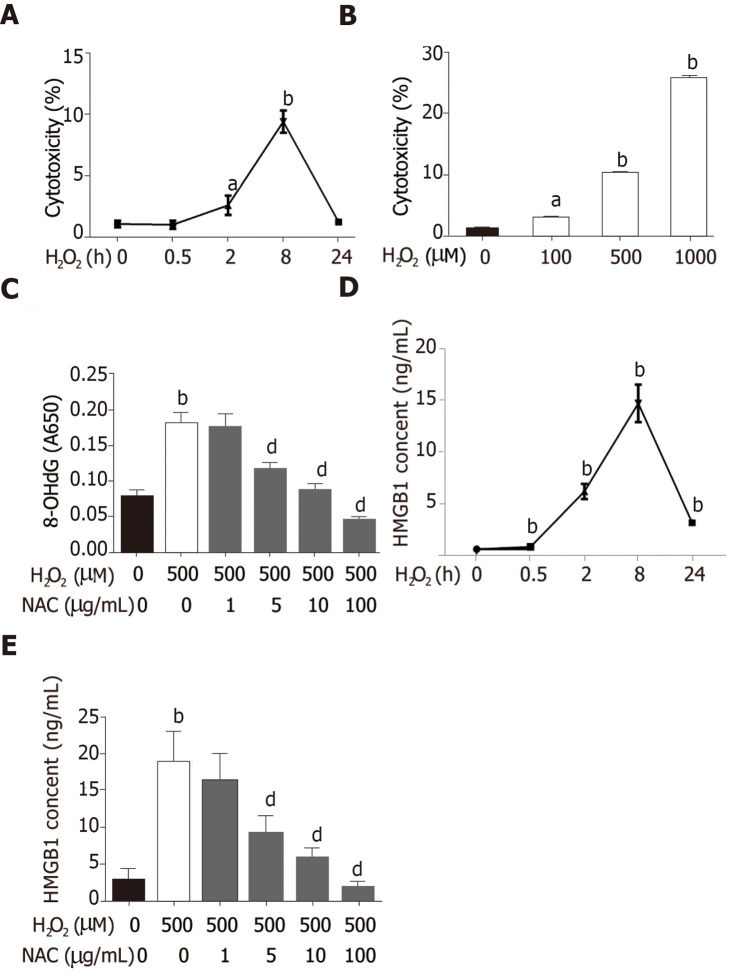
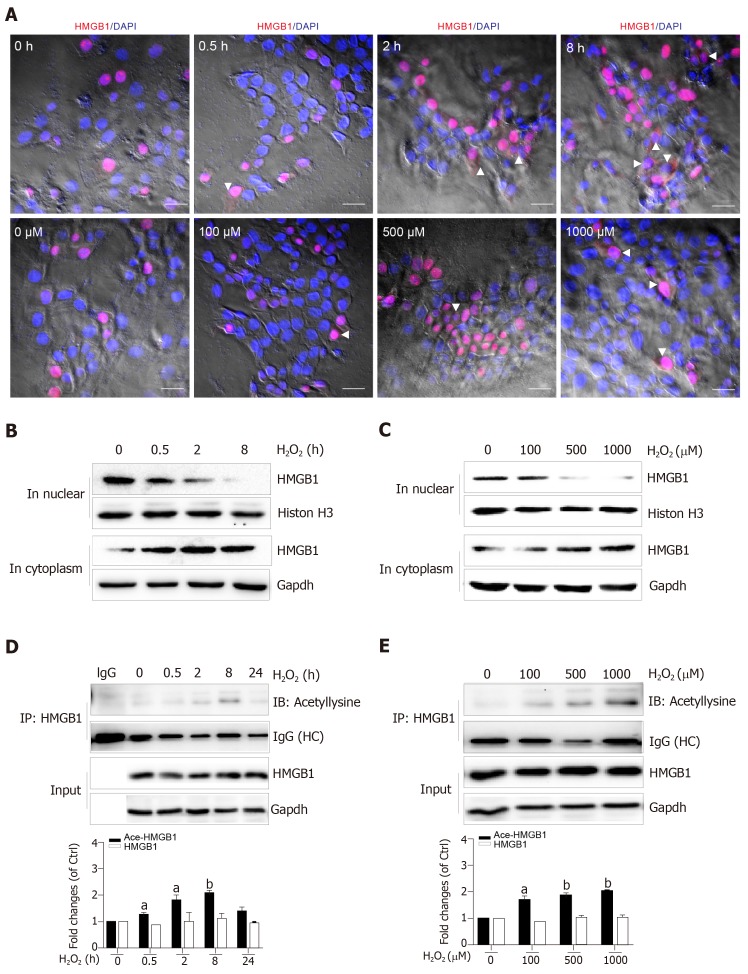
C57BL6/J male mice were fed a high-fat diet for 12 wk plus a single binge of ethanol to induce severe steatohepatitis. Hepatocytes treated with HO were used to establish an in vitro model. Serum alanine aminotransferase, liver HO content and catalase activity, lactate dehydrogenase and 8-hydroxy-2-deoxyguanosine content, nicotinamide adenine dinucleotide (NAD) levels, and Sirtuin 1 (Sirt1) activity were detected by spectrophotometry. HMGB1 release was measured by enzyme linked immunosorbent assay. HMGB1 translocation was observed by immunohistochemistry/immunofluorescence or Western blot. Relative mRNA levels were assayed by qPCR and protein expression was detected by Western blot. Acetylated HMGB1 and poly(ADP-ribose)polymerase 1 (Parp1) were analyzed by Immunoprecipitation.
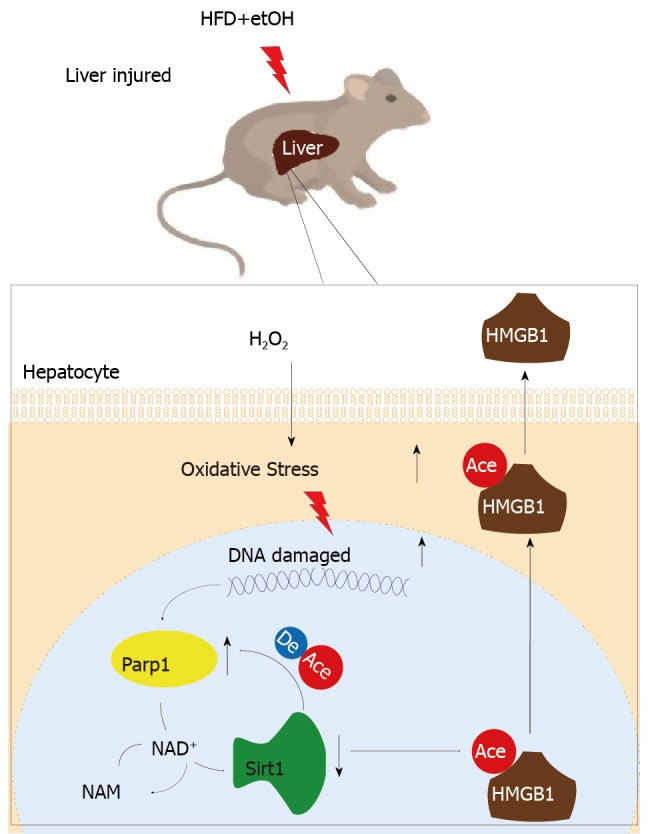
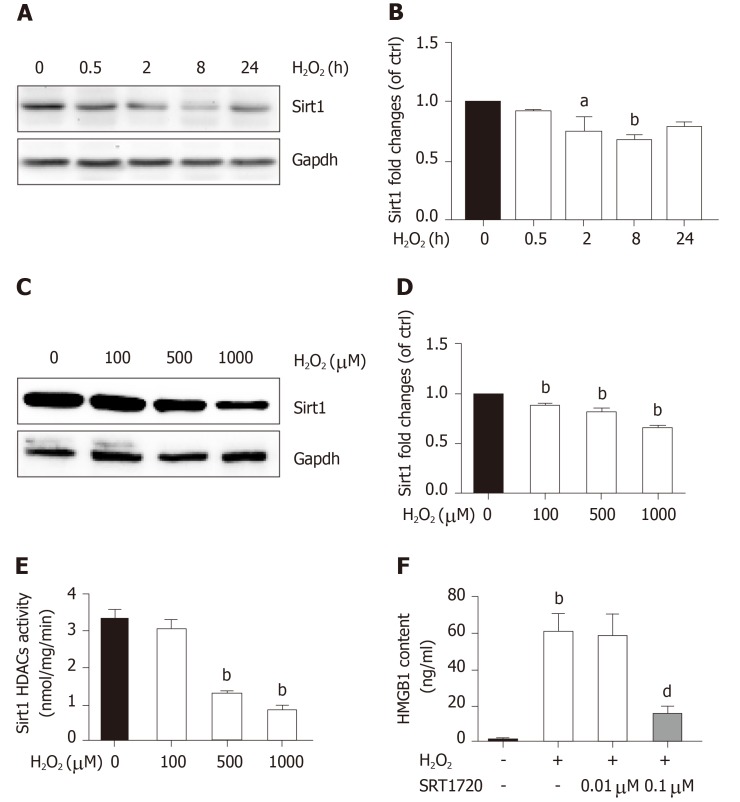
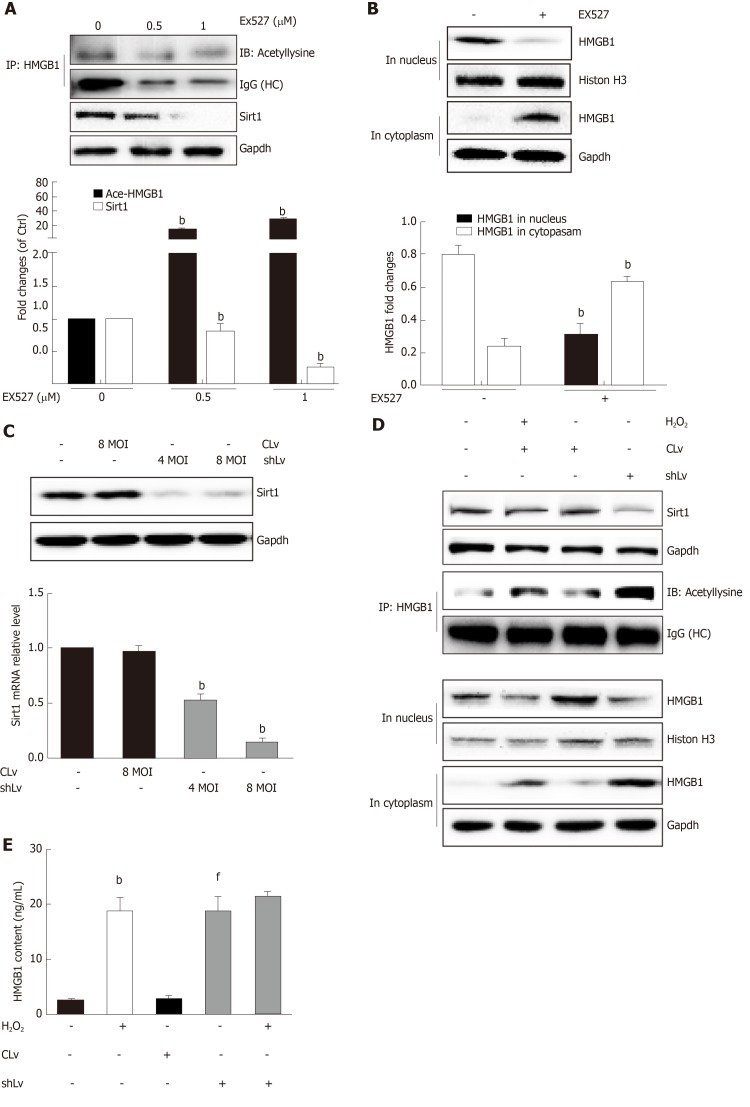
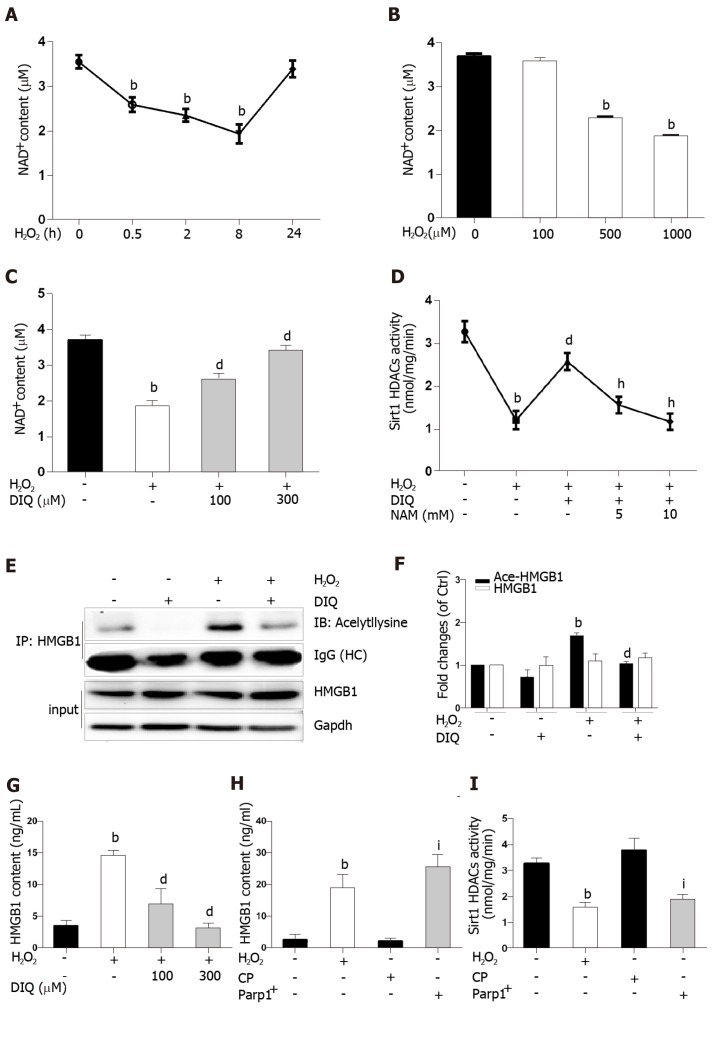
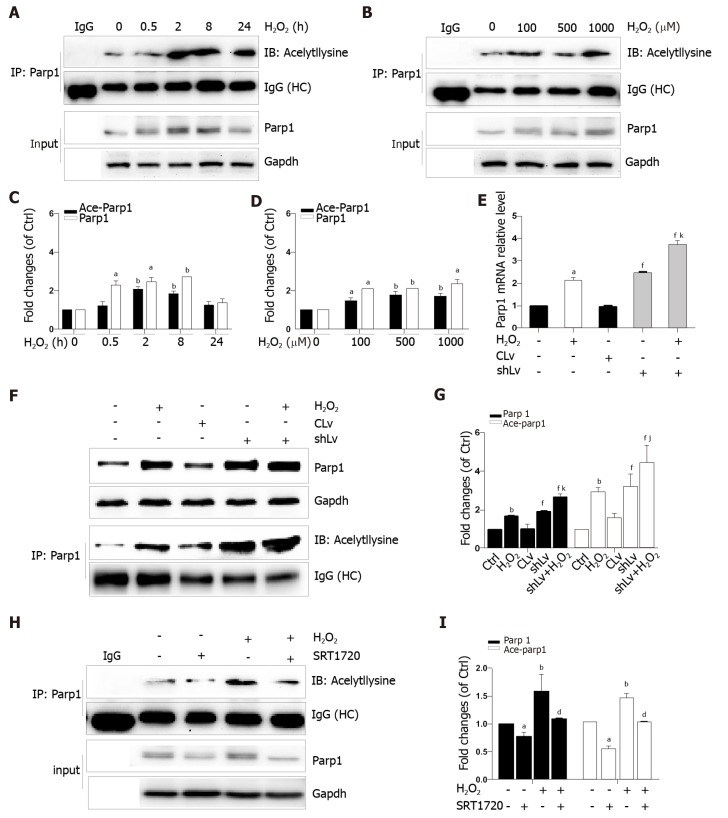
When hepatocytes were damaged, HMGB1 translocated from the nucleus to the cytoplasm because of its hyperacetylation and was passively released outside both in vivo and in vitro. After treatment with Sirt1-siRNA or Sirt1 inhibitor (EX527), the hyperacetylated HMGB1 in hepatocytes increased, and Sirt1 activity inhibited by HO could be reversed by Parp1 inhibitor (DIQ). Parp1 and Sirt1 are two NAD-dependent enzymes which play major roles in the decision of a cell to live or die in the context of stress . We showed that NAD depletion attributed to Parp1 activation after DNA damage was caused by oxidative stress in hepatocytes and resulted in Sirt1 activity inhibition. On the contrary, Sirt1 suppressed Parp1 by negatively regulating its gene expression and deacetylation.
The functional inhibition between Parp1 and Sirt1 leads to HMGB1 hyperacetylation, which leads to its translocation from the nucleus to the cytoplasm and finally outside the cell.
高迁移率族蛋白 B1(HMGB1)被认为是损伤相关分子模式的代表,在细胞损伤/死亡时释放,引发炎症反应,最终导致组织损伤。数十项研究表明,HMGB1 参与了某些疾病,但关于受损肝细胞如何释放 HMGB1 的细节仍需进一步研究。
揭示氧化应激状态下肝细胞中 HMGB1 的释放机制。
雄性 C57BL6/J 小鼠给予高脂饮食 12 周,同时单次给予乙醇灌胃诱导严重脂肪性肝炎。用 HO 处理的肝细胞建立体外模型。通过分光光度法检测血清丙氨酸氨基转移酶、肝 HO 含量和过氧化氢酶活性、乳酸脱氢酶和 8-羟基-2-脱氧鸟苷含量、烟酰胺腺嘌呤二核苷酸(NAD)水平和 Sirtuin 1(Sirt1)活性。通过酶联免疫吸附试验检测 HMGB1 释放。通过免疫组化/免疫荧光或 Western blot 观察 HMGB1 易位。通过 qPCR 检测相对 mRNA 水平,通过 Western blot 检测蛋白表达。通过免疫沉淀分析乙酰化 HMGB1 和聚(ADP-核糖)聚合酶 1(Parp1)。
当肝细胞受损时,HMGB1 因 hyperacetylation 从细胞核易位到细胞质,并在体内和体外被动释放到细胞外。用 Sirt1-siRNA 或 Sirt1 抑制剂(EX527)处理后,肝细胞中 hyperacetylated HMGB1 增加,HO 抑制的 Sirt1 活性可被 Parp1 抑制剂(DIQ)逆转。Parp1 和 Sirt1 是两种 NAD 依赖性酶,在应激条件下对细胞是生存还是死亡的决策中起主要作用。我们表明,由于氧化应激引起的肝细胞 DNA 损伤导致的 NAD 耗竭会导致 Parp1 激活,从而抑制 Sirt1 活性。相反,Sirt1 通过负调控其基因表达和去乙酰化来抑制 Parp1。
Parp1 和 Sirt1 之间的功能抑制导致 HMGB1 hyperacetylation,导致其从细胞核易位到细胞质,最终到细胞外。