Moran Robert F, McKay David, Tornstrom Paulynne C, Aziz Alex, Fernandes Arantxa, Grau-Crespo Ricardo, Ashbrook Sharon E
School of Chemistry, EaStCHEM and Centre of Magnetic Resonance , University of St Andrews , St Andrews KY16 9ST , U.K.
Department of Chemistry , University of Reading , Whiteknights RG6 6AD , U.K.
J Am Chem Soc. 2019 Nov 6;141(44):17838-17846. doi: 10.1021/jacs.9b09036. Epub 2019 Oct 22.
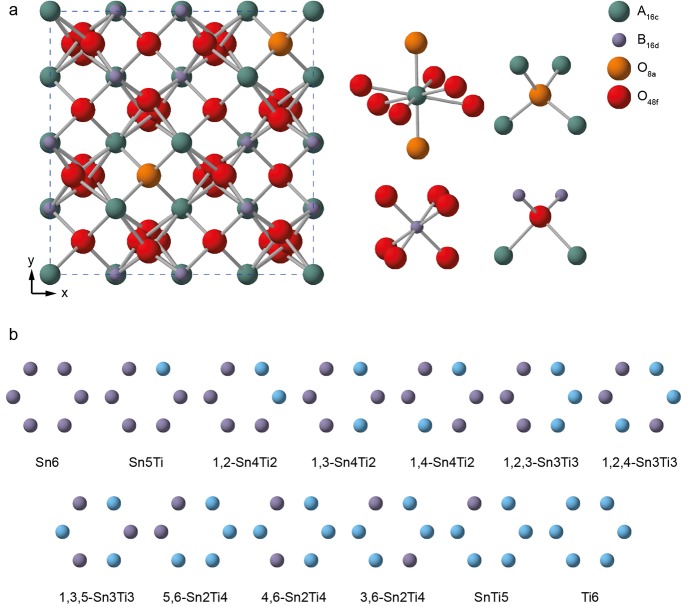
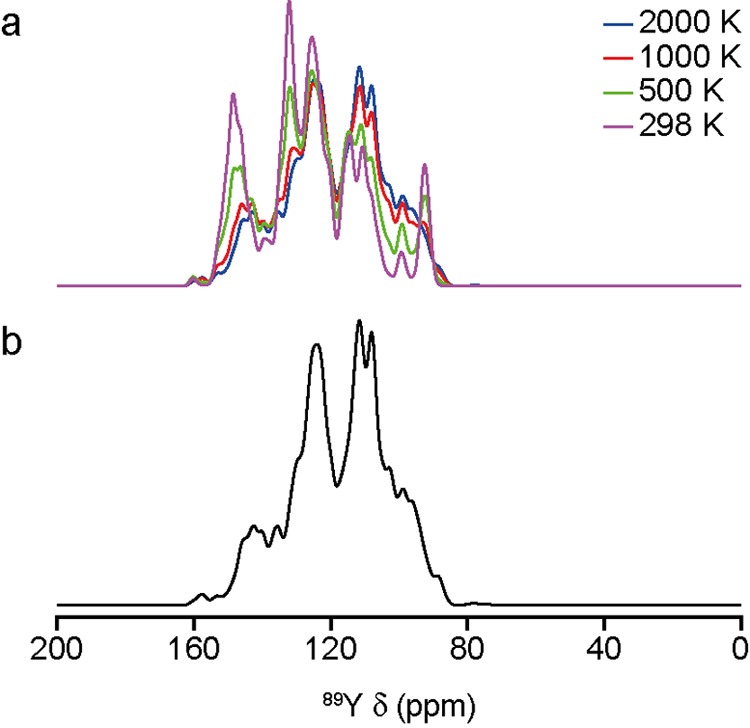
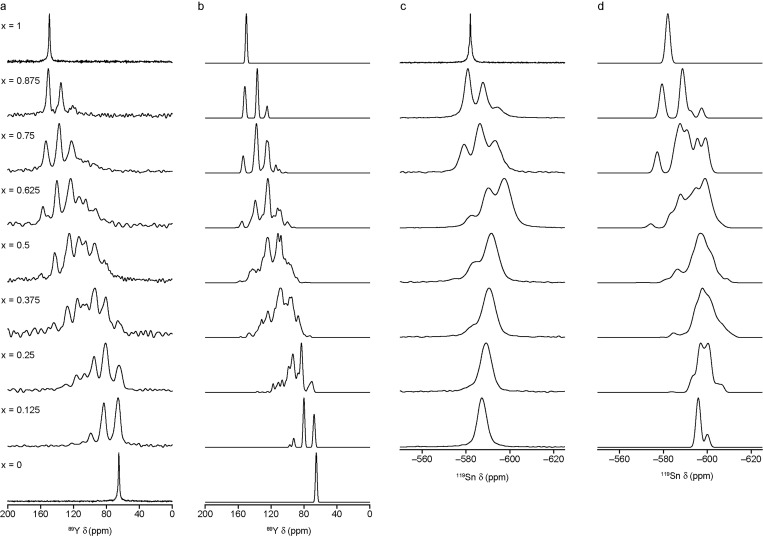
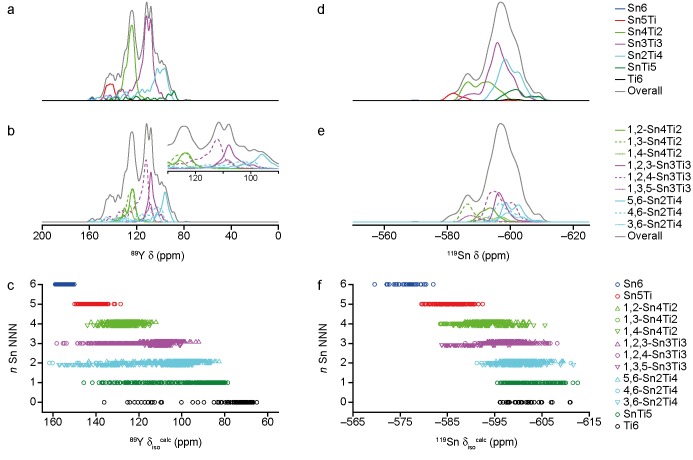
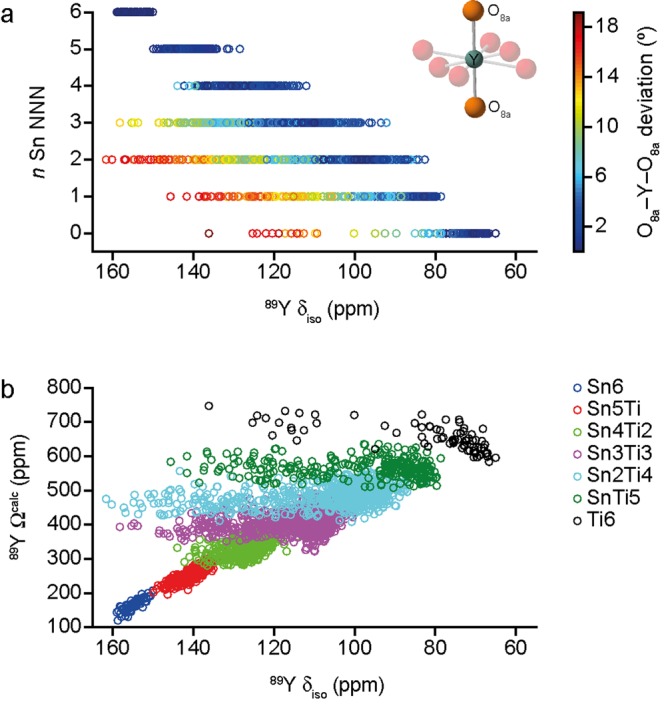
The sensitivity of NMR to the local environment, without the need for any long-range order, makes it an ideal tool for the characterization of disordered materials. Computational prediction of NMR parameters can be of considerable help in the interpretation and assignment of NMR spectra of solids, but the statistical representation of all possible chemical environments for a solid solution is challenging. Here, we illustrate the use of a symmetry-adapted configurational ensemble in the simulation of NMR spectra, in combination with solid-state NMR experiments. We show that for interpretation of the complex and overlapped lineshapes that are typically observed, it is important to go beyond a single-configuration representation or a simple enumeration of local environments. The ensemble method leads to excellent agreement between simulated and experimental spectra for Y(Sn,Ti)O pyrochlore ceramics, where the overlap of signals from different local environments prevents a simple decomposition of the experimental spectral lineshapes. The inclusion of a Boltzmann weighting confirms that the best agreement with experiment is obtained at higher temperatures, in the limit of full disorder. We also show that to improve agreement with experiment, in particular at low dopant concentrations, larger supercells are needed, which might require alternative simulation approaches as the complexity of the system increases. It is clear that ensemble-based modeling approaches in conjunction with NMR spectroscopy offer great potential for understanding configurational disorder, ultimately aiding the future design of functional materials.
核磁共振对局部环境的敏感性,无需任何长程有序结构,使其成为表征无序材料的理想工具。核磁共振参数的计算预测对解释和归属固体核磁共振谱有很大帮助,但对固溶体所有可能化学环境的统计表示具有挑战性。在此,我们展示了结合固态核磁共振实验,使用对称适配构型系综来模拟核磁共振谱。我们表明,对于解释通常观察到的复杂且重叠的线形,超越单构型表示或简单列举局部环境很重要。对于Y(Sn,Ti)O烧绿石陶瓷,系综方法使模拟谱与实验谱高度吻合,不同局部环境的信号重叠使得实验谱线形难以简单分解。包含玻尔兹曼权重证实,在较高温度下,即完全无序的极限情况下,与实验的吻合度最佳。我们还表明,为了提高与实验的吻合度,特别是在低掺杂浓度下,需要更大的超胞,随着系统复杂性增加,这可能需要采用替代模拟方法。显然,基于系综的建模方法与核磁共振光谱相结合,在理解构型无序方面具有巨大潜力,最终有助于功能材料的未来设计。