Stanford University, Department of Electrical Engineering, Stanford, 94305, USA.
University of Illinois Urbana-Champaign, Department of Electrical and Computer Engineering, Urbana, 61801, USA.
Sci Rep. 2019 Oct 21;9(1):15067. doi: 10.1038/s41598-019-51418-z.
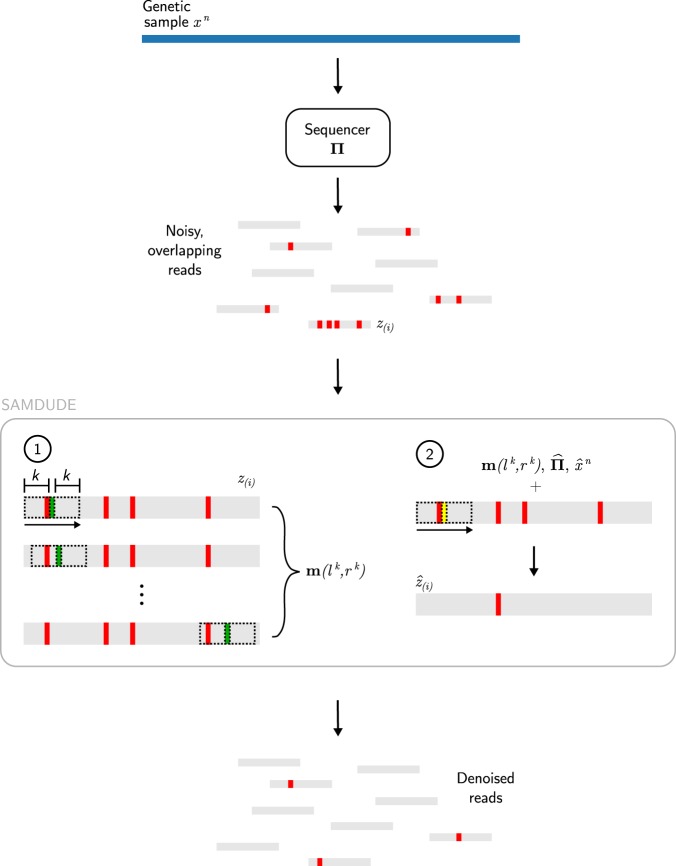
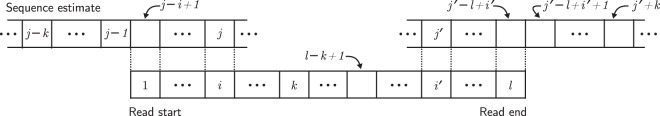
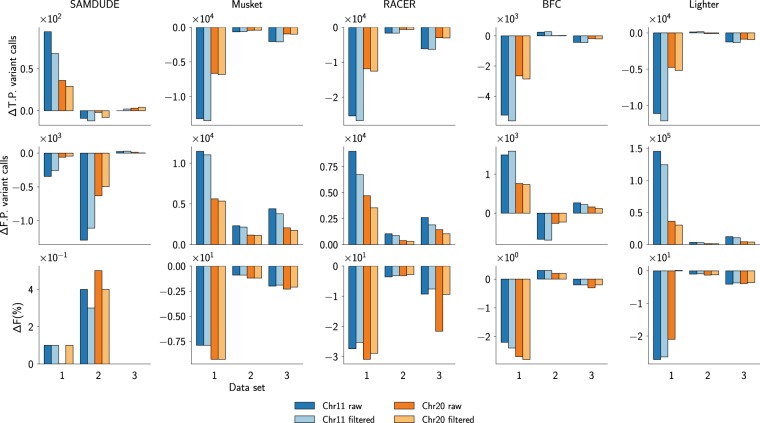
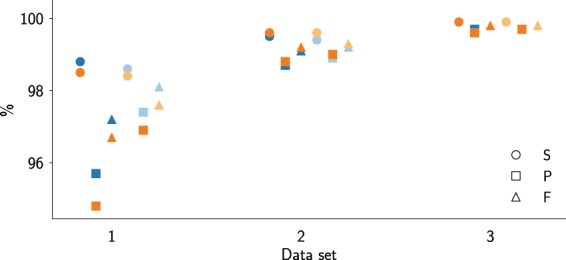
Noise in genomic sequencing data is known to have effects on various stages of genomic data analysis pipelines. Variant identification is an important step of many of these pipelines, and is increasingly being used in clinical settings to aid medical practices. We propose a denoising method, dubbed SAMDUDE, which operates on aligned genomic data in order to improve variant calling performance. Denoising human data with SAMDUDE resulted in improved variant identification in both individual chromosome as well as whole genome sequencing (WGS) data sets. In the WGS data set, denoising led to identification of almost 2,000 additional true variants, and elimination of over 1,500 erroneously identified variants. In contrast, we found that denoising with other state-of-the-art denoisers significantly worsens variant calling performance. SAMDUDE is written in Python and is freely available at https://github.com/ihwang/SAMDUDE .
基因组测序数据中的噪声已知会对基因组数据分析管道的各个阶段产生影响。变异识别是这些管道中的许多步骤的重要步骤,并且越来越多地用于临床环境中以辅助医疗实践。我们提出了一种去噪方法,称为 SAMDUDE,它在对齐的基因组数据上运行,以提高变异调用性能。用 SAMDUDE 对人类数据进行去噪导致个体染色体和全基因组测序 (WGS) 数据集的变异识别得到改善。在 WGS 数据集,去噪导致鉴定出近 2000 个额外的真实变异,消除了 1500 多个错误识别的变异。相比之下,我们发现用其他最先进的去噪器进行去噪会显著降低变异调用性能。SAMDUDE 是用 Python 编写的,可以在 https://github.com/ihwang/SAMDUDE 上免费获得。