Wellcome-MRC Cambridge Stem Cell Institute, University of Cambridge, Cambridge CB2 0AW, UK.
Computational Biology Group, Department of Molecular Biology, Max Planck Institute for Developmental Biology, Max-Planck Ring 1, 72076 Tübingen, Germany.
Nucleic Acids Res. 2021 Aug 20;49(14):e83. doi: 10.1093/nar/gkab433.
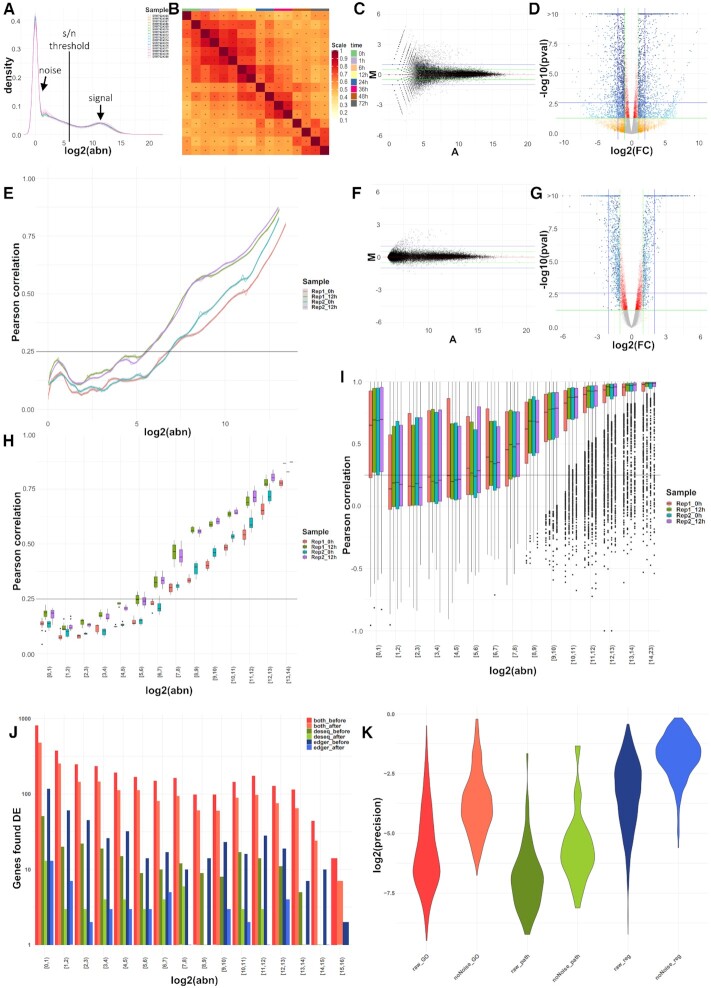
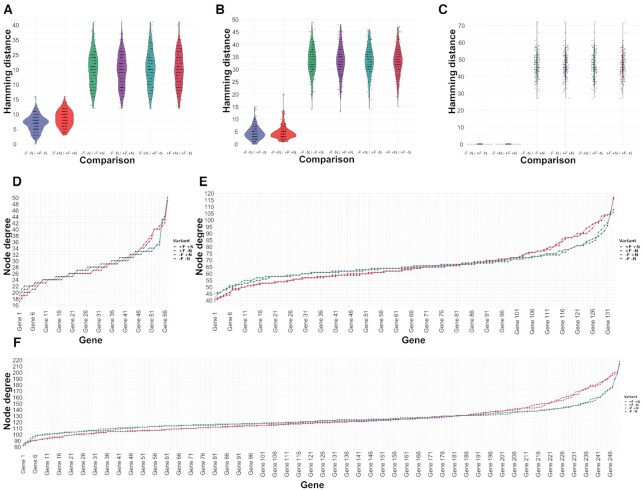
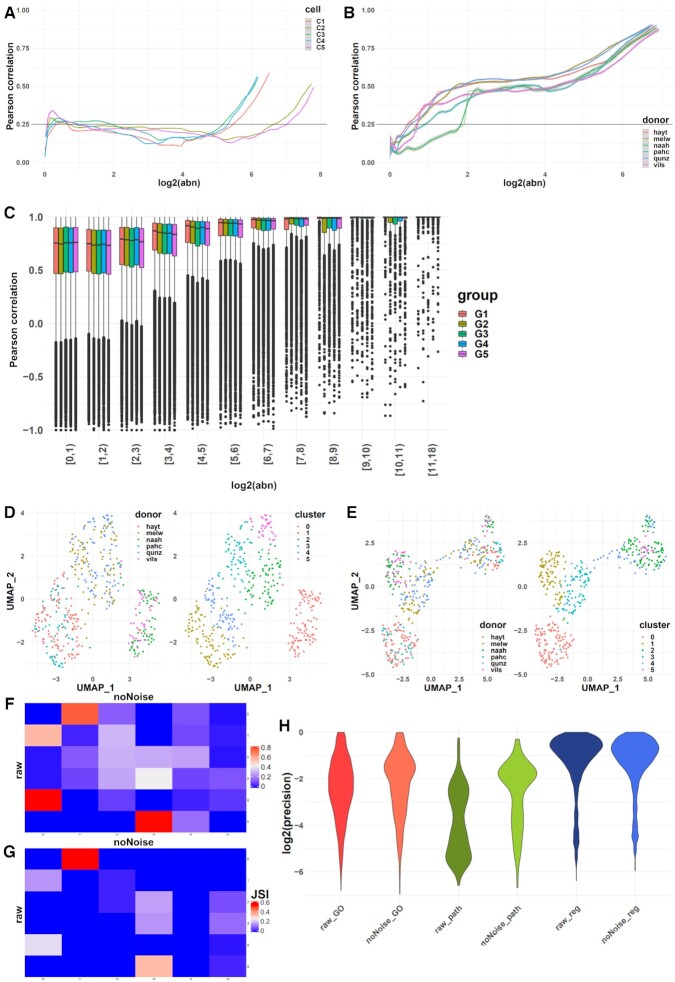
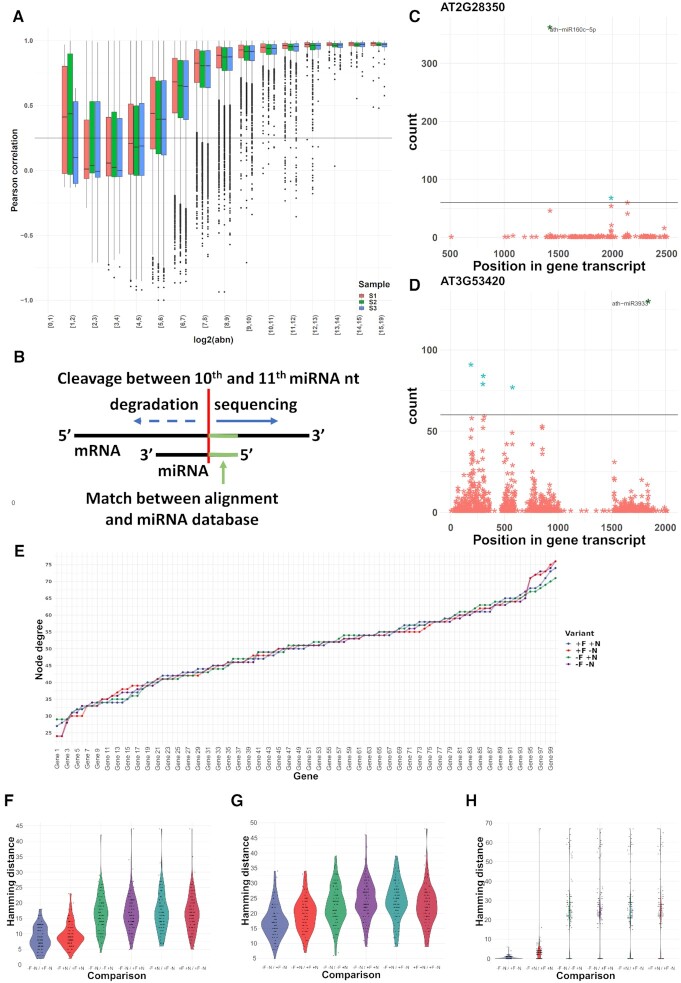
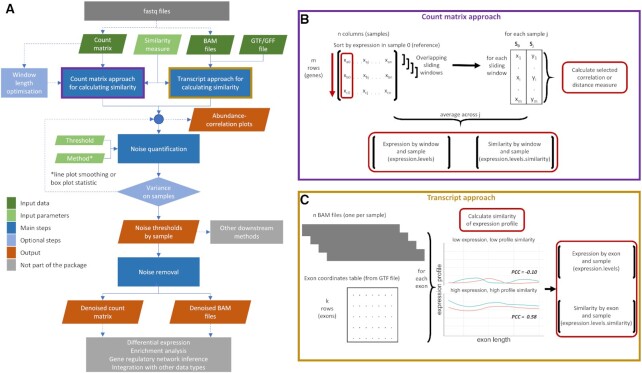
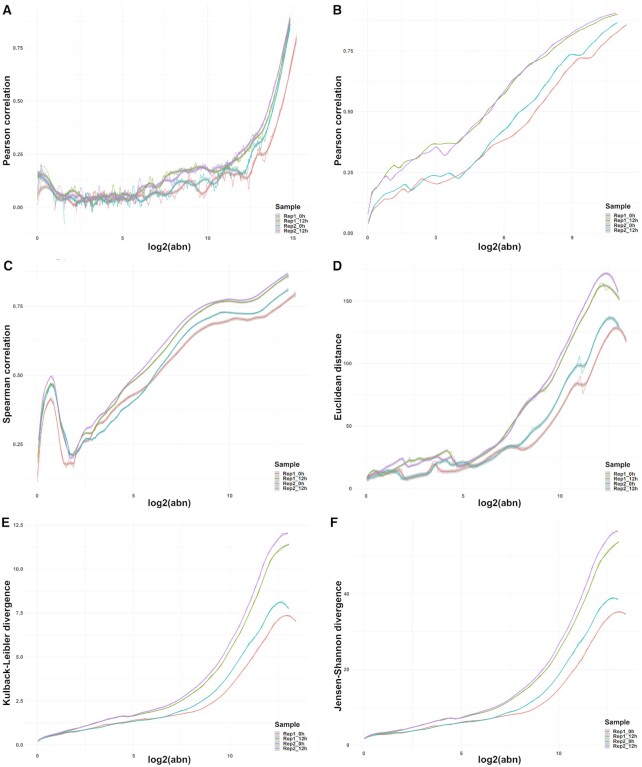
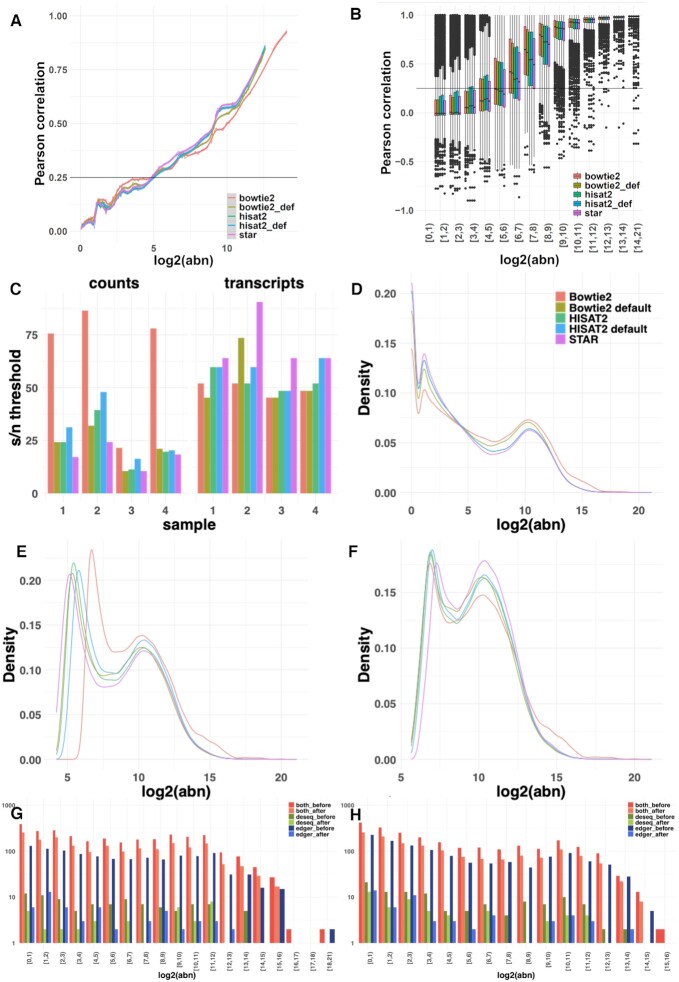
High-throughput sequencing enables an unprecedented resolution in transcript quantification, at the cost of magnifying the impact of technical noise. The consistent reduction of random background noise to capture functionally meaningful biological signals is still challenging. Intrinsic sequencing variability introducing low-level expression variations can obscure patterns in downstream analyses. We introduce noisyR, a comprehensive noise filter to assess the variation in signal distribution and achieve an optimal information-consistency across replicates and samples; this selection also facilitates meaningful pattern recognition outside the background-noise range. noisyR is applicable to count matrices and sequencing data; it outputs sample-specific signal/noise thresholds and filtered expression matrices. We exemplify the effects of minimizing technical noise on several datasets, across various sequencing assays: coding, non-coding RNAs and interactions, at bulk and single-cell level. An immediate consequence of filtering out noise is the convergence of predictions (differential-expression calls, enrichment analyses and inference of gene regulatory networks) across different approaches.
高通量测序能够以前所未有的分辨率进行转录本定量,但代价是放大了技术噪声的影响。一致地降低随机背景噪声以捕捉具有功能意义的生物学信号仍然具有挑战性。内在测序变异性引入的低水平表达变化可能会使下游分析中的模式变得模糊。我们引入了 noisyR,这是一种全面的噪声滤波器,用于评估信号分布的变化,并在重复和样本之间实现最佳的信息一致性;这种选择还促进了背景噪声范围之外有意义的模式识别。noisyR 适用于计数矩阵和测序数据;它输出特定于样本的信号/噪声阈值和过滤后的表达矩阵。我们举例说明了在各种测序检测中,在各种数据集上最小化技术噪声的影响:在批量和单细胞水平上的编码、非编码 RNA 和相互作用。过滤掉噪声的一个直接后果是不同方法之间的预测(差异表达调用、富集分析和基因调控网络推断)趋于一致。